Design, Expression, and Inhibitory Effects of Antagonistic Single-Chain Platelet-Derived Growth Factor on Lung Cancer Cells
2025-01-23 | Volume 3 Issue 1 - Volume 3 | Research Articles | Salim AlhafyanAbstract
Platelet-derived growth factor (PDGF) plays a pivotal role in numerous physiological and pathological conditions, acting as a dimeric disulfide-bound protein that induces receptor dimerization and subsequently activates cell signaling pathways. Given its involvement in various malignant and benign diseases, targeting the PDGF signaling pathway is a promising therapeutic strategy. In this study, a single-chain antagonist PDGF (sc-PDGF) was designed by leveraging structural insights from the PDGF BB/receptor complex (PDB ID: 3JMG). The design involved substituting a receptor-binding site in one pole while keeping the other intact. Molecular dynamics simulations of the designed antagonistic sc-PDGF showed a noteworthy reduction in binding affinity at the mutant interface, indicating a reduced capability to effectively bind the receptor. The antagonistic sc-PDGF was successfully expressed in Escherichia coli, purified, and refolded using a one-step affinity chromatography approach. Far-UV circular dichroism spectroscopy demonstrated no significant alterations in the secondary structure of the designed sc-PDGF. Additionally, MTT assays confirmed the inhibitory effect of the sc-PDGF on lung cancer cells (A549), with an IC50 value of 0.72 µg/ml. This result underscores the potential of the sc-PDGF antagonist as a viable drug candidate for suppressing the proliferation of cancer cells, particularly those in which PDGF plays a critical role in cell growth.
Keywords : Platelet-Derived Growth Factor; PDGF; Antagonist; Cancer; Molecular Dynamics Simulation; Receptor Dimerization; Escherichia Coli Expression; MTT Assay.
INTRODUCTION
Growth factors assume crucial roles in regulating cell proliferation, growth, and differentiation under both physiological and pathological conditions. One such pivotal factor is the platelet-derived growth factor (PDGF), which participates in various physiological activities. PDGF contributes to the differentiation of embryonic organs, facilitates wound healing processes, regulates interstitial pressure within tissues, and plays a key role in platelet aggregation. The multifaceted involvement of PDGF underscores its significance in maintaining homeostasis and responding to dynamic cellular processes in health and disease (1, 2). PDGF is a dimeric polypeptide, each monomer weighing approximately 30 kDa and consisting of nearly 100 amino acid residues. Five isoforms of PDGF exist, denoted as AA, BB, CC, DD, and AB. These isoforms act as activators of the PDGF receptor (PDGFR), which is present in two isoforms, PDGFR-α and PDGFR-β. The activation process involves receptor homo- or hetero-dimerization, leading to the induction of autophosphorylation on specific tyrosine residues located within the inner side of the receptor. This autophosphorylation event triggers the activation of kinase activity, initiating the phosphorylation of downstream proteins (3). The ensuing phosphorylation cascade orchestrates the effects of the PDGF signaling pathway (4). PDGF is involved in a number of malignant and benign diseases, including glioblastoma multiforme (GBM) (5), meningiomas, chordoma, and ependymoma (6, 7). Additionally, PDGF plays a role in skin cancer, specifically dermatofibrosarcoma protuberans (DFSP) (8), gastrointestinal tumors (GIST), synovial sarcoma, osteosarcoma , hepatocellular carcinoma, and prostate cancer (3, 9). Aberrantly elevated levels of PDGF receptor and/or PDGF have been observed in lymphomas and leukemias, including chronic myelogenous leukemia (CML) (10), acute lymphoblastic leukemia (ALL), chronic eosinophilic leukemia (CEL), and anaplastic large cell lymphoma (11, 12). Moreover, such abnormal upregulation has been noted in other cancer types, such as breast carcinoma, sarcomatoid non-small-cell lung cancer, and colorectal cancer (13). These findings underscore the potential role of dysregulated PDGF signaling in the pathogenesis of these hematologic and solid malignancies, suggesting its relevance as a target for further therapeutic exploration. PDGF exerts its influence not only in malignant diseases, but also in non-malignant conditions, extending its influence to fibrotic diseases such as kidney, liver, cardiac, and lung fibrosis. Additionally, PDGF plays a role in various vascular disorders, including systemic sclerosis, pulmonary arterial hypertension (PAH), endothelial barrier dysfunction, proliferative retinopathy, cerebral vasospasm, and cytomegalovirus infection (14, 15). The broad spectrum of PDGF involvement highlights its significance in the context of diverse pathological processes, emphasizing its potential as a therapeutic target in addressing both malignant and non-malignant disorders. Inhibition of the PDGF signaling pathway holds significant therapeutic potential for both malignant and non-malignant diseases. Various strategies have been devised to impede this pathway, including the utilization of monoclonal antibodies targeting PDGF or PDGFR. These antibodies specifically obstruct the PDGF signaling pathway by binding to PDGF or PDGFR, thereby preventing receptor dimerization (16, 17). Alternatively, small molecule inhibitors of receptor kinases present another strategy, although they may lack specificity and inadvertently inhibit other signaling pathways (18). Another approach involves the use of soluble receptors that compete with PDGFR for binding to the ligand, thereby preventing the interaction between PDGF and its receptor. Furthermore, DNA aptamers, oligonucleotides that bind to PDGF and hinder its interaction with its own receptor, represent an additional avenue for therapeutic intervention (19). These diverse strategies offer a range of options for modulating the PDGF signaling pathway with the aim of treating various diseases. Imatinib, a tyrosine kinase inhibitor that effectively impedes the PDGF pathway, has been approved for the treatment of chronic myelogenous leukemia (CML), acute lymphoblastic leukemia (ALL), chronic eosinophilic leukemia (CEL), gastrointestinal stromal tumors (GIST), and dermatofibrosarcoma protuberans (DFSP). Another PDGFR-selective inhibitor, CP-673451, has demonstrated inhibitory effects on the proliferation and migration of lung cancer cells. Moreover, CP-673451 has exhibited the capacity to enhance the cytotoxicity of cisplatin and induce apoptosis in non-small cell lung cancer (20). In a phase II trial, Olaratumab®— (a human anti-PDGFR-α monoclonal antibody)-displayed an acceptable safety profile in patients with metastatic gastrointestinal stromal tumors (21). These instances underscore the therapeutic potential of targeting the PDGF pathway for the treatment of various malignancies. In this investigation, we focused on the design and construction of a single-chain PDGF receptor antagonist. This antagonist was strategically engineered such that one of its two poles retained the capability to bind with the receptor while the other pole lacked this ability. The intended outcome was to impede receptor dimerization, thereby inhibiting the PDGF signaling pathway. This inhibitory effect is achieved by displacing specific amino acid residues within PDGF BB that play a crucial role in the interaction between PDGF and its receptor. This targeted interference was informed by a meticulous analysis of the structure of the receptor-ligand complex, as illustrated in Scheme 1.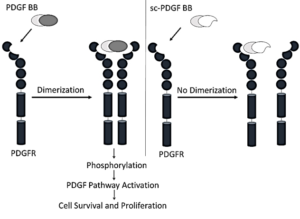
Scheme 1. Mechanism of Designed Single-Chain Antagonistic PDGF, which binds to one monomer PDGFR, prevents dimerization of receptors and therefore inhibits the PDGF signaling pathway.
MATERIALS AND METHODS
Materials
Isopropyl β-D-1-thiogalactopyranoside (IPTG) and kanamycin were procured from Invitrogen (Carlsbad, CA, USA). Nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography resin was supplied by Qiagen (Hilden, Germany). A 96-Well plate, specifically Max-iSorp, was provided by Nunc (USA). Oxidized glutathione was purchased from AppliChem (USA), while reduced glutathione was acquired from BioBasic (Canada). (3-(4,5-dimethyl thiazolyl-2)-2,5-diphenyltetrazolium bromide) MTT was obtained from Sigma (USA). Escherichia coli strain BL21 (DE3) was procured from Novagen (Madison, WI, USA) and New England Biolabs Inc. (Beverly, MA, USA), respectively. Cell culture medium was sourced from Bioidea Company (Tehran, Iran), and fetal bovine serum was acquired from Gibco/Invitrogen (Carlsbad, CA, USA). A549 cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). All other chemicals used in the study were obtained from Merck (Darmstadt, Germany). YASARA software version 14.12.2 was employed for visualizing protein figures.
Design of PDGF Antagonist
The crystal structures of the PDGF-PDGF Receptor complex (PDB ID: 3MJG) were obtained from the Protein Data Bank (PDB), ensuring a reliable foundation for subsequent analyses. The CFinder server (http://bioinf.modares.ac.ir/software/nccfinder/) was used to identify the residues that are critical in the interaction between PDGF and PDGFR that cause receptor dimerization.
Residue Analysis and Replacement Strategy
For a detailed exploration of the residues engaged in protein-protein interactions within the PDGF-PDGF Receptor complex, the CFinder server was employed. This computational tool utilizes the protein complex PDB file as input, relying on accessible surface area differences (delta-ASA) to identify residues that contribute to ligand-receptor interactions. Subsequently, to facilitate the replacement of PDGF segments involved in binding to PDGFR, peptide segments with comparable geometry but distinct physicochemical properties were selected.
Protein Design and Fragment Replacement Strategy
The ProDA (Protein Design Assistant) server (http://bioinf.modares.ac.ir/software/proda) was instrumental in this process (22). This server aids in the identification of peptide segments suitable for substitution, ensuring the maintenance of structural integrity while introducing variations in physicochemical characteristics. This integrated approach, combining CFinder and ProDA servers, enhances our understanding of the intricate molecular interactions within the PDGF-PDGF Receptor complex and guides the design of the single-chain PDGF receptor antagonist with targeted modifications for disrupting receptor dimerization. The ProDA (Protein Design Assistant) web server, integral to our study, provides a comprehensive list of diverse protein segments by querying a database using specified input parameters. The criteria employed in the search encompass the number of amino acid residues, amino acid sequence patterns, secondary structure, distance between fragment ends, as well as the polarity and accessibility patterns of amino acid residues. The selection of suitable fragments is meticulously carried out based on several considerations, including amino acid content and specific characteristics such as secondary structure features, polarity, and accessibility patterns. These selected fragments from the candidate sequences are then strategically chosen for replacement within the PDGF BB sequence. This sophisticated approach, combining criteria-driven segment selection with subsequent integration into the PDGF BB sequence, ensures a thoughtful and targeted modification strategy in the design of our single-chain PDGF receptor antagonist.
Linker Design and 3D Structure Construction
To optimize purification and refolding processes while minimizing interference with the three-dimensional structure of the single-chain PDGF (sc-PDGF), an 18-amino acid residue linker was meticulously designed. This linker serves as a critical bridge between the two monomers of PDGF BB. Subsequently, the three-dimensional structure of the modified PDGF was constructed based on its primary sequence. The MODELLER software (version 9.17) (23) was employed for this purpose, generating a pool of 100 models. The model selection process involved choosing the model with the lowest MODELLER objective function score, indicating the best structural fit. To ensure the structural integrity and quality of the selected model, stereochemistry checks were performed using PROCHECK software (24). This rigorous validation step guarantees the reliability and accuracy of the constructed 3D structure, which is essential for subsequent analyses and experimental applications.
Molecular Dynamics Simulations
Molecular dynamics (MD) simulations were conducted employing GROMACS 5.0.7, focusing on both the modeled single-chain PDGF (sc-PDGF) and the native isoform. The simulations spanned a duration of 20 nanoseconds, utilizing the Gromos96 force field (25). The structure was solvated in a solvation box using a simple point-charge water model (26), with a minimum distance of 10 Å between the protein and the edges of the box. The system was neutralized by adding Cl– and Na+ ions that were randomly replaced with water molecules. The system was initially relaxed, and any bad contacts between atoms were removed through the steepest descent algorithm in an energy minimization step. The minimized systems were then equilibrated for 100 picoseconds (ps) using canonical and isothermal–obaric ensembles. The simulations were performed at 300 K and 1 bar. Finally, the equilibrated systems were simulated for a period of 20 nanoseconds (ns) with a 2-femtosecond (fs) time step to determine the possible effects of modification on the structure of sc-PDGF. The Root Mean Square Deviation (RMSD) and radius of gyration of the system were investigated and evaluated to determine the stability of the MD simulations and the compactness of the sc-PDGF during the simulations.
Molecular Docking Analysis
To assess the binding capabilities of both the native and modified PDGF with PDGFR, molecular docking simulations were conducted using the ClusPro server (https://cluspro.org) (27). Molecular Docking was performed with a monomeric receptor, and the ability of native/ modified PDGF to bind to the receptor was evaluated depending on the ClusPro score, and the results of Docking were evaluated.
Construction, Expression, Refolding, and Purification of Antagonistic PDGF
The PDGF antagonist-encoding gene was synthesized and subsequently cloned into the pET28a expression vector, flanked by BamHI/XhoI restriction sites. This molecular construct was facilitated by Shine Gene Molecular Biotech, Inc. (Shanghai, China). The steps involved in the construction, expression, refolding, and purification of the PDGF antagonist are detailed below:
Gene Cloning and Transformation
The synthesized PDGF antagonist-encoding gene was cloned into the pET28a expression vector, which was then transformed into Escherichia coli BL21 (DE3) cells.
Expression Conditions
The transformed cells were induced for expression at 37 °C, with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 6 hours.
Inclusion Body Collection and Dissolution
Inclusion bodies containing the expressed PDGF antagonist were collected and dissolved in 6 M urea.
Purification and Refolding
Purification and refolding were conducted using a previously described protocol (28). Column chromatography was employed with sequential elution using buffers A, B, C, D, and E respectively;
Buffers A: 6 mol/L urea, 0.5 mol/L NaCl, 10% glycerol, 1% TritonX-100, 20 mM Tris, pH 6.5.
Buffers B: 6 mol/L urea, 0.5 mol/L NaCl, 10% glycerol, 1% TritonX-100, 20 mM Tris, pH 5.8.
Buffers C: 4 mol/L urea, 0.5 mol/L NaCl, 6% glycerol, 20 mM Tris, 2 mM reduced glutathione (GSH), pH 8.0.
Buffers D: 2 mol/L urea, 0.5 mol/L NaCl, 3% glycerol, 20 mM Tris, 2 mM GSH, 0.2 mM oxidized glutathione (GSSG), pH 8.0.
Buffers E: 0.5 mol/L NaCl, 20 mM Tris, 2 mM GSH, 0.5 mM GSSG, pH 8.0. The elution buffer contained 300 mM imidazole and 0.5 mol/L NaCl.
Elution and Gel Analysis
The eluted modified PDGF was collected in sterile vials. The collected fractions were loaded onto an electrophoresis gel for further analysis. This detailed procedure outlines the steps taken to construct, express, and purify the modified PDGF antagonist, ensuring its structural integrity and functionality for subsequent experiments.
Circular Dichroism Measurement
CD spectra were performed using a spectropolarimeter (Jasco J-715, Japan) at the far-UV wavelength of 195-240 nm (sc-PDGF concentration was 0.1 mg/ml in phosphate saline buffer), to confirm that the secondary structures of refolded sc-PDGF were not significantly changed. The data were smoothed by the Jasco J-715 software to reduce the routine noise and calculate the secondary structure percentage of antagonistic PDGF. The results were reported as molar ellipticity [θ] (deg cm2.dmol-1), based on a mean amino acid residue weight (MRW) of sc-PDGF. The content of secondary structures of sc-PDGF was obtained and compared to those of modeled sc-PDGF and the crystal structure of native PDGF BB.
Growth Inhibition Assay
The inhibitory activity of modified PDGF was studied on adenocarcinomic human alveolar basal epithelial cells (A549). The cells were cultured in DMEM with 10% FBS and incubated in 5% CO2 at 37 °C. For growth inhibition assay, cells were collected by washing with PBS, added trypsin, then counted and 6000 cells/well were seeded in a sterile 96-well plate. After 24 h, the medium was replaced with fresh medium containing different concentrations of modified PDGF. The cells were incubated for 24 h at 37 °C. Afterward, cell growth inhibition was analyzed using the MTT assay. 10 µl of 5 mg/ml MTT solution was added to each well and the plates were incubated for 3-4 h at 37 °C. After that, the media were replaced with 100 µl of DMSO (dimethyl sulfoxide), and the absorbance of the wells was measured at 570 nm using a µQuant microplate reader (BioTek, USA) (29).
RESULTS
- Designing of PDGF Antagonist
The design of the PDGF antagonist was informed by an analysis of accessible surface area (ASA) differences, identifying critical peptide segments and amino acid residues in PDGF BB involved in receptor binding. The fragments exhibiting the highest delta-ASA were recognized as crucial in the binding process to the receptor. Specifically:
1. In PDGF (subunit I): Fragments 13IAE15, 54NNRN57, and 98KCET101 were identified as essential for binding to the receptor (Figure 1A).
- In PDGF (subunit II): Fragments 27RRLIDRTNANFLVW40, and 77IVRLLPIF84 were recognized as critical for binding to the receptor (Figure 1B).
Based on these findings, strategic replacements were decided upon in four important regions (Figure 1C):
- E15 with K in PDGFB monomer I
- Fragment (54 NNRN 57) with (ADED) in PDGFB monomer I
- Fragment 25-43 changed to LIRPPIC in PDGFB monomer II
- Fragment 74-85 changed to KLDGAK in PDGFB monomer II (Table 1).
In the design of the single-chain PDGF (sc-PDGF), a linker sequence VGSTSGSGKSSEGKGEVV was incorporated. This linker serves to connect the C-terminus of subunit I of PDGF BB to the N-terminus of subunit II. The construction of the designed sc-PDGF was executed using MODELLER, and the best structure was meticulously selected for further analyses (Figure 1D). This refined sc-PDGF structure incorporates strategic modifications and a linker sequence to enhance its functional properties, setting the stage for subsequent evaluations. Figure 1. PDGF BB binding sites determined by C Finder. Critical amino acid residues in the binding receptor in subunit I (A) and II (B) of native PDGF, 3D structure of native PDGF BB (C), and 3D structure of single-chain PDGF (D). The candidate binding sites to be modified, the substituted amino acid residues, and the linker are shown in red, green and yellow, respectively.
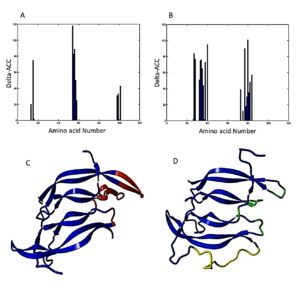
Table 1. PDGF BB fragments are supposed to be modified, and fragments that replace them have similar geometry and secondary structure but different physicochemical properties.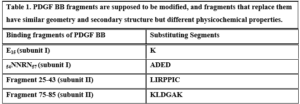
- Molecular Dynamics Simulations
The 3D structure of the designed single-chain PDGF (sc-PDGF) was modeled based on the crystal structure of wild-type PDGF BB. The structure with the lowest MODELLER objective function was selected for molecular dynamics (MD) simulations. The objectives of the MD simulations were to refine the sc-PDGF structures under similar conditions, compare them with native PDGF BB, and allow conformational relaxation before the docking study. After the simulations, the Root Mean Square Deviation (RMSD) and radius of gyration values for the backbone atoms of sc-PDGF were monitored relative to the starting structure during the MD production phase. The RMSD curves (Figure 2) indicated that the backbone atoms of the sc-PDGF structures were stable and reached equilibrium after 10 ns of simulation. Both structures exhibited RMSD values with no significant deviation. Additionally, the radius of gyration for the modeled sc-PDGF during the simulations showed negligible changes, indicating minimal alterations in the compactness of the proteins (Figure 2). These results affirm the stability and structural integrity of the modeled sc-PDGF during MD simulations, providing a solid foundation for subsequent analyses.
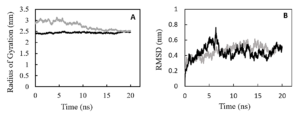
Figure 2. Molecular dynamic simulations result, RMSD and radius of gyration of the proteins during the simulations. RMSD (A) and radius of gyration (B) values of the backbone atoms of native PDGF BB (black) and sc-PDGF (gray) structures with respect to the reference coordinate during 20ns simulations.
- Molecular Docking
The binding ability of the modified PDGF to the receptors was predicted using ClusPro and compared with native PDGF. The docking results revealed distinctive features between native PDGF and the modified PDGF: Native PDGF demonstrated two high-score positions capable of binding to PDGF receptors (PDGFRs). These positions were located on two symmetrical binding sites at its two poles. The modified PDGF exhibited only one high-score position, aligning with the anticipated outcome. As expected, the modified interface of sc-PDGF lost its ability to bind the receptor, and the modified PDGF could only bind to PDGFR through one pole with a high score. Consequently, the dimerization of receptors cannot take place (Figure 3).
Table 2. Protein-Protein Interaction Prediction by ClusPro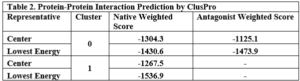 These results from ClusPro, as summarized in Table 2, confirm the differential binding scores and binding sites between native PDGF and the modified sc-PDGF. In Cluster 0, both native PDGF and modified PDGF show high scores for binding to one pole, with the intended binding site for sc-PDGF. In Cluster 1, native PDGF exhibits a high score for the symmetrical pole, while the modified PDGF shows a low score, indicating altered binding characteristics. The antagonistic sc-PDGF does not display any binding on the modified pole, supporting its role in preventing receptor dimerization.
These results from ClusPro, as summarized in Table 2, confirm the differential binding scores and binding sites between native PDGF and the modified sc-PDGF. In Cluster 0, both native PDGF and modified PDGF show high scores for binding to one pole, with the intended binding site for sc-PDGF. In Cluster 1, native PDGF exhibits a high score for the symmetrical pole, while the modified PDGF shows a low score, indicating altered binding characteristics. The antagonistic sc-PDGF does not display any binding on the modified pole, supporting its role in preventing receptor dimerization.
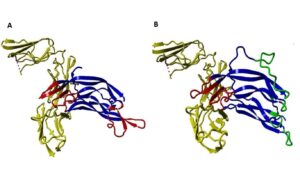
Figure 3. Protein-Protein Docking results ClusPro. The interaction between native PDGF BB and PDGFR. Native PDGF BB can bind with the receptor (yellow) by its own two equal poles shown in red (A), and the interaction between sc-PDGF and PDGFR, can only bind with the receptor by its unchanged pole (red sites). Substituted fragments cannot bind to the receptor, shown in green (B).
- Construction of Active Antagonistic PDGF
The synthesis and expression of the modified PDGF-encoded gene were carried out in E. coli BL21 (DE3). Subsequent steps in the construction of active antagonistic PDGF involved the collection and washing of insoluble inclusion bodies with plate wash buffer. The inclusion bodies were then dissolved using a solution buffer, filtered through a 0.22 μm filter, and loaded onto a Ni-NTA agarose column. Purification and refolding processes were performed concurrently on the column, and finally, 0.5 ml eluted samples were collected.
The success of the purification process was confirmed through SDS-PAGE analysis, as depicted in Figure 4.
Figure 4. SDS-PAGE analysis of the expressed and purified sc-PDGF. Inclusion body in protein expression obtained from E. coli BL21 (DE3) (A) and SDS-PAGE results of refolding and purification on Ni-NTA affinity chromatography column. Lanes 1-4, eluted fractions collected from Ni-NTA affinity column (B).
- Calculation of Secondary Structure Contents of sc-PDGF using CD Spectrum
The secondary structure contents of the single-chain PDGF (sc-PDGF) were calculated using the CD spectrum and compared to the predicted model and the crystal native structure. The results, as presented in Table 3, indicate slight differences between the calculated secondary structure contents of sc-PDGF and the predicted model and crystal native structure.
Table 3. Secondary structure contents of sc-PDGF obtained by CD compared to predicted from modeled sc-PDGF and crystal PDGF BB 3D structure.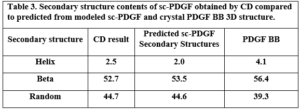 Anti-proliferation effect of Antagonistic PDGF
Anti-proliferation effect of Antagonistic PDGF
A cell viability test was conducted using A549 cells to assess the inhibitory effect of modified PDGF. The experiment involved incubating and treating 6000 A549 cells with different concentrations of the modified PDGF (Figure 5). The results indicate a dose-dependent inhibitory effect on cell proliferation; At a concentration of 0.25 μg/ml of PDGF antagonist, there was approximately a 30% inhibition of A549 cell proliferation compared to the control; a concentration of 0.75 μg/ml of PDGF antagonist resulted in approximately a 50% inhibition of cell growth; the highest concentration tested, 3 μg/ml of PDGF antagonist, resulted in a remarkable inhibition of cell proliferation, reaching up to about 90%. The concentration that inhibits 50% of cell proliferation (IC50) was calculated using Prism software and found to be 0.7151 μg/ml (27.7 nM).
Figure 5. Anti-proliferation Effect of Antagonistic PDGF on A549 Cells. Each concentration was performed with 3 replicates, error bar ≈ ± SD (standard deviation).
These results demonstrate the potent anti-proliferative activity of the modified PDGF antagonist on A549 lung cancer cells, indicating its potential as a therapeutic agent for inhibiting cancer cell growth.
DISCUSSION
The inhibition of the platelet-derived growth factor (PDGF) signaling pathway has been identified as a crucial target for the treatment of various malignant and nonmalignant diseases, including cancer and fibrotic diseases, where PDGF plays a pivotal role. The selective inhibition of the PDGF signaling pathway offers numerous advantages in the treatment of diverse diseases, minimizing potential side effects on other cells (16). In this study, we focused on designing and constructing a single-chain PDGF receptor antagonist, aiming to disrupt the dimerization of PDGF receptors and subsequently inhibit the PDGF signaling pathway. This approach is significant given the central role of PDGF in physiological and pathological conditions. The designed single-chain antagonistic PDGF (sc-PDGF) was constructed based on structural information derived from the PDGF BB/receptor complex. Molecular dynamics simulations and structural analyses were employed to evaluate the binding affinity and stability of the sc-PDGF mutant interface. The successful expression, purification, and refolding of sc-PDGF were confirmed through various techniques, including far-UV CD spectroscopy. The molecular docking results showed that sc-PDGF had a reduced ability to bind to PDGF receptors compared to native PDGF, supporting its potential as an effective antagonist. The calculated secondary structure contents of sc-PDGF, obtained through CD spectroscopy, indicated minimal changes, further affirming the structural integrity of the designed antagonist. Furthermore, the anti-proliferation assay demonstrated the potent inhibitory effect of sc-PDGF on A549 lung cancer cells in a dose-dependent manner. The calculated IC50 value highlighted the concentration at which 50% of cell proliferation was inhibited. This study provides valuable insights into the development of a targeted therapeutic approach for diseases associated with aberrant PDGF signaling. The designed sc-PDGF shows promise as a selective antagonist with potential applications in the treatment of cancer and fibrotic diseases, offering a novel avenue for the development of targeted therapies with minimized off-target effects. Future investigations may focus on in vivo studies and clinical applications to validate the therapeutic efficacy of the designed sc-PDGF. Current PDGF antagonists, particularly small molecule kinase inhibitors such as Imatinib, exhibit non-selectivity, leading to undesired side effects on various tissues (3, 29). Additionally, antibodies, while effective, come with high costs and may stimulate the immune system, posing potential challenges (29, 30). In our research, we aimed to develop a selective PDGF antagonist. The PDGF signaling pathway is initiated by the dimerization of PDGF receptors through dimeric PDGF. In our study, we focused on modifying one pole of the PDGF dimer, allowing the antagonistic PDGF to bind exclusively to one receptor. This modification prevents receptor dimerization, subsequently selectively inhibiting the PDGF signaling pathway. In a similar approach, Ghavami et al. successfully designed and synthesized a potent VEGF antagonist capable of inhibiting angiogenesis and preventing capillary tube formation in HUVEC cell lines (31). This strategy of selectively targeting specific pathways by modifying critical interaction sites has shown promise in controlling pathological processes. Our engineered sc-PDGF antagonist, designed to disrupt the dimerization of PDGF receptors, holds the potential for selective inhibition of the PDGF signaling pathway. This approach provides a novel alternative to existing PDGF antagonists, addressing issues related to non-selectivity and cost associated with current therapeutic options. Further studies, including in vivo investigations and clinical trials, will be crucial to validate the therapeutic efficacy and safety profile of the designed sc-PDGF. We identified the crucial amino acid residues responsible for binding to the receptor at one pole of PDGF BB within the shared interface of two subunits (Figure 1). Subsequently, we modified these residues to hinder binding, specifically -replacing Glu15 with Lys, introducing an opposite charge. We replaced the segment 54NNRN57 with ADED, which has opposite physicochemical properties while maintaining the same geometry. Additionally, the two crucial binding fragments, 25-43 and 74-85, in the other subunit were replaced with two turns. These turns were carefully selected from a database to ensure that they maintained the original geometry without amino acid residues that bind to the receptor (Table 1). The PDGF BB isoform was chosen due to its ability to bind and activate all PDGF receptor types (αα, ββ, and the heterodimer complex αβ). Furthermore, the crystal structure of the PDGF BB/PDGFR complex has been elucidated. We determined the sequence of the engineered sc-PDGF antagonist and modeled its 3D structure (Figure 1D). Molecular dynamics simulations were conducted on the modeled sc-PDGF to facilitate the conformational relaxation of its structure before the docking study. The RMSD and radius of gyration values indicated stable behavior with no significant deviation, as illustrated in Figure 2. Furthermore, the docking binding scores of both native and modified sc-PDGF to the receptor indicate a noteworthy difference. The native PDGF exhibits two high-scoring positions precisely on the expected sites, whereas the sc-PDGF shows only one high-scoring position (Table 2 and Figure 3). This suggests that the modified interface may have lost its ability to effectively bind to the receptor. The coding sequences of the sc-PDGF gene were synthesized and incorporated into pET28a expression vectors. Subsequently, E. coli BL21 (DE3) was transformed, and the modified sc-PDGF was expressed and refolded as outlined in the methods section. The presence of a linker between two PDGF monomers and a His tag at the N-terminus facilitated the purification and refolding process, streamlined by the Ni-NTA affinity chromatography column, as illustrated in Figure 4. The designed PDGF antagonist exhibited inhibitory effects on A549 cell proliferation, with a concentration of 3 µg/ml causing a notable reduction in cell growth to 10% compared to the control (Figure 5). This observation underscores the antagonist’s inhibitory impact on PDGFR, achieved through the prevention of receptor dimerization. Furthermore, the modified pole of sc-PDGF lost its ability to bind to the receptor, confirming the intended impact. According to the ClusPro docking results, sc-PDGF is predicted to have lost the ability to bind to two receptor molecules simultaneously. This loss is crucial in the context of PDGF dimerization and signaling, aligning with the findings from MTT assays. The results confirm the inhibitory effect of the antagonistic sc-PDGF on A549 cell lines, which is consistent with previous research. In a related study, demonstrated that inhibiting the PDGF receptor can effectively suppress cell growth in the A549 cell line (32). Our study’s notable advantage lies in the extracellular mechanism of inhibition, which has the potential to prevent cellular uptake. This approach addresses the challenges associated with cellular uptake, as well as intracellular metabolism and degradation of the drug (33, 34). Furthermore, the high selectivity of the designed antagonistic PDGF suggests a potential reduction in side effects on other cells. In a related study, Boesen et al. [reference] prepared single-chain variants of VEGF by incorporating a 14-residue linker between two monomers. Their findings demonstrated that these single-chain variants were fully functional and equivalent to the wild-type VEGF. In their work, Zhao et al. also successfully prepared an effective single-chain antagonist of VEGF (35). This was achieved by deleting and substituting critical binding site residues in one monomer of the native VEGF while keeping the other monomer intact. This strategic modification prevented the dimerization of the receptors, consequently inhibiting the VEGF signaling pathway (35). In parallel studies, Khafaga et al. and Qin et al. designed antagonistic VEGF variants by structurally analyzing VEGF and modifying amino acid residues at the binding site on one pole of the protein (36, 37). They successfully produced antagonistic single-chain VEGF and confirmed its inhibitory effect. Additionally, Kassem et al. demonstrated the antagonization of growth hormone (GH) by preventing receptor dimerization (38). This was achieved through the binding of one receptor molecule by monovalent fragments of GH, effectively preventing receptor dimerization and inhibiting the signaling pathway (39). Activation of PDGF receptors, similar to VEGF and growth hormone receptors, necessitates binding of ligands at two distinct sites to initiate receptor dimerization. Consequently, by deleting or modifying one binding site while preserving the other, the ligand occupies only one receptor, preventing the dimerization of receptors. This strategic modification inhibits the cascade phosphorylation of the receptor and its subsequent effects. In conclusion, PDGF signaling inhibitors have demonstrated efficacy in various clinical applications, particularly in certain cancers and fibrotic diseases. The engineered sc-PDGF antagonist, designed to bind to a single receptor, effectively prevents the dimerization of PDGFRs and inhibits their signaling pathway. Docking results highlighted the inability of the modified PDGF to bind on one pole while retaining binding on the other. The proliferation assay confirmed the inhibitory effects on A549 cells, suggesting that the sc-PDGF antagonist could serve as a potential therapeutic agent for diseases involving the PDGF signaling pathway.
References :- Heldin CH, Lennartsson J, Westermark B. Involvement of platelet‐derived growth factor ligands and receptors in tumorigenesis. Journal of internal medicine. 2018;283(1):16-44.
- Magar AG, Morya VK, Kwak MK, Oh JU, Noh KC. A Molecular Perspective on HIF-1α and Angiogenic Stimulator Networks and Their Role in Solid Tumors: An Update. International Journal of Molecular Sciences. 2024;25(6):3313.
- Ai J-Y, Liu C-F, Zhang W, Rao G-W. Current status of drugs targeting PDGF/PDGFR. Drug Discovery Today. 2024:103989.
- Olsen RS, Dimberg J, Geffers R, Wågsäter D. Possible role and therapeutic target of PDGF-D signalling in colorectal cancer. Cancer Investigation. 2019;37(2):99-112.
- Simpson JE, Muir MT, Lee M, Naughton C, Gilbert N, Pollard SM, Gammoh N. Autophagy supports PDGFRA-dependent brain tumor development by enhancing oncogenic signaling. Developmental Cell. 2024;59(2):228-43. e7.
- Tamura R, Sato M, Morimoto Y, Ohara K, Kosugi K, Oishi Y, et al. Quantitative assessment and clinical relevance of VEGFRs-positive tumor cells in refractory brain tumors. Experimental and Molecular Pathology. 2020;114:104408.
- Saratsis AM, Knowles T, Petrovic A, Nazarian J. H3K27M mutant glioma: disease definition and biological underpinnings. Neuro-oncology. 2024;26(Supplement_2):S92-S100.
- Singh H, Choudhary HB, Mandlik DS, Magre MS, Mohanto S, Ahmed MG, et al. Molecular pathways and therapeutic strategies in dermatofibrosarcoma protuberans (DFSP): unravelling the tumor’s genetic landscape. EXCLI Journal. 2024;23:727-62.
- Chen H, Sha N, Liu N, Hu H. Effects of bone marrow mesenchymal stem cell-conditioned medium on the proliferation and migration of liposarcoma cells. Folia Histochemica et Cytobiologica. 2024;62(1):50-60.
- Hussain S, Mursal M, Verma G, Hasan SM, Khan MF. Targeting oncogenic kinases: Insights on FDA approved tyrosine kinase inhibitors. European Journal of Pharmacology. 2024:176484.
- Miyauchi J. The hematopoietic microenvironment of the fetal liver and transient abnormal myelopoiesis associated with Down syndrome: A review. Critical Reviews in Oncology/Hematology. 2024:104382.
- Lin L-H, Lin J-S, Yang C-C, Cheng H-W, Chang K-W, Liu C-J. Overexpression of platelet-derived growth factor and its receptor are correlated with oral tumorigenesis and poor prognosis in oral squamous cell carcinoma. International journal of molecular sciences. 2020;21(7):2360.
- Carvalho I, Milanezi F, Martins A, Reis RM, Schmitt F. Overexpression of platelet-derived growth factor receptor α in breast cancer is associated with tumour progression. Breast Cancer Research. 2005;7:1-8.
- Varol A, Klauck SM, Dantzer F, Efferth T. Enhancing Cisplatin Drug Sensitivity through PARP3 Inhibition: The Influence on PDGF and G-Coupled Signal Pathways in Cancer. Chemico-Biological Interactions. 2024:111094.
- Strell C, Östman A. Stromal PDGF Receptors; Impact on Prognosis and Response to Treatment. Biomarkers of the Tumor Microenvironment: Springer; 2022. p. 125-38.
- Pandey P, Khan F, Upadhyay TK, Seungjoon M, Park MN, Kim B. New insights about the PDGF/PDGFR signaling pathway as a promising target to develop cancer therapeutic strategies. Biomedicine & Pharmacotherapy. 2023;161:114491.
- Zou X, Tang X-Y, Qu Z-Y, Sun Z-W, Ji C-F, Li Y-J, Guo S-D. Targeting the PDGF/PDGFR signaling pathway for cancer therapy: A review. International Journal of Biological Macromolecules. 2022;202:539-57.
- Song C-Y, Chang SL-Y, Lin C-Y, Tsai C-H, Yang S-Y, Fong Y-C, et al. Visfatin-Induced Inhibition of miR-1264 Facilitates PDGF-C Synthesis in Chondrosarcoma Cells and Enhances Endothelial Progenitor Cell Angiogenesis. Cells. 2022;11(21):3470.
- Sae-Lim S, Soontornworajit B, Pichayanoot P. Inhibition of colorectal cancer cell proliferation by regulating platelet-derived growth factor B signaling with a DNA aptamer. Asian Pacific Journal of Cancer Prevention: APJCP. 2019;20(2):487.
- Yang Y, Deng Y, Chen X, Zhang J, Chen Y, Li H, et al. Inhibition of PDGFR by CP-673451 induces apoptosis and increases cisplatin cytotoxicity in NSCLC cells via inhibiting the Nrf2-mediated defense mechanism. Toxicology Letters. 2018;295:88-98.
- Xing T, Zhang Y, Li X, Guo M, Liang W, He J. Response to the combination use of pazopanib with olaratumab in a patient with lung-metastatic embryonal rhabdomyosarcoma: a case report. Translational Lung Cancer Research. 2021;10(1):483.
- Shirvanizadeh N, Vriend G, Arab SS. Loop modelling 1.0. Journal of Molecular Graphics and Modelling. 2018;84:64-8.
- Webb B, Sali A. Comparative protein structure modeling using MODELLER. Current protocols in bioinformatics. 2016;54(1):5.6. 1-5.6. 37.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of applied crystallography. 1993;26(2):283-91.
- Scott WR, Hünenberger PH, Tironi IG, Mark AE, Billeter SR, Fennen J, et al. The GROMOS biomolecular simulation program package. The Journal of Physical Chemistry A. 1999;103(19):3596-607.
- Berendsen HJ, Postma JP, van Gunsteren WF, Hermans J, editors. Interaction models for water in relation to protein hydration. Intermolecular forces: proceedings of the fourteenth Jerusalem symposium on quantum chemistry and biochemistry held in jerusalem, israel, april 13–16, 1981; 1981: Springer.
- Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, et al. The ClusPro web server for protein–protein docking. Nature protocols. 2017;12(2):255-78.
- Luo W-j, Zhao G-Q, Wang C-f, Wang L-m, Wang X-j. Cloning, expression and functional analyses of human platelet-derived growth factor-B chain peptide for wound repair of cat corneal endothelial cells. Chinese Journal of Traumatology. 2009;12(01):31-7.
- Erdoğan Ü, Erbaş S, Muhammed MT, Onem E, Soyocak A, Ak A. Isolation and characterization of thymoquinone from Nigella sativa essential oil: antioxidant and antibacterial activities, molecular modeling studies, and cytotoxic effects on lung cancer A549 cells. Journal of Essential Oil Bearing Plants. 2024:1-17.
- Costa A, Scalzulli E, Carmosino I, Ielo C, Bisegna ML, Martelli M, Breccia M. Pharmacotherapeutic advances for chronic myelogenous leukemia: beyond tyrosine kinase inhibitors. Expert Opinion on Pharmacotherapy. 2024;25(2):189-202.
- Ghavamipour F, Shahangian SS, Sajedi RH, Arab SS, Mansouri K, Aghamaali MR. Development of a highly‐potent anti‐angiogenic VEGF 8–109 heterodimer by directed blocking of its VEGFR‐2 binding site. The FEBS journal. 2014;281(19):4479-94.
- Huber L, Birk R, Rotter N, Aderhold C, Lammert A, Jungbauer F, Kramer B. Effect of small-molecule tyrosine kinase inhibitors on PDGF-AA/BB and PDGFRα/β expression in SCC according to HPV16 status. Anticancer Research. 2020;40(2):825-35.
- Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine & growth factor reviews. 2004;15(4):197-204.
- Sadeghi-Ardebili M, Hasannia S, Dabirmanesh B, Khavari-Nejad RA. Functional characterization of the dimeric form of PDGF-derived fusion peptide fabricated based on theoretical arguments. Scientific Reports. 2024;14(1):1003.
- Li M, Zhao X, Wu G, Wang W, Du J, Xu G, et al. Using capillary electrophoresis sodium dodecyl sulfate (CE‐SDS) and liquid chromatograph mass spectrometry (LC–MS) to identify glycosylated heavy chain heterogeneity in the anti‐VEGFR‐2 monoclonal antibody. Electrophoresis. 2024.
- Khafaga AF, Gaballa MM, Karam R, Shoulah SA, Shamma RN, Khalifa NE, et al. Synergistic therapeutic strategies and engineered nanoparticles for anti-vascular endothelial growth factor therapy in cancer. Life Sciences. 2024:122499.
- Qin J, Qu S, Zhu K, Cheng Y, Pan G, Jing W, et al. Rational design and synthesis of 6-aryl-6H-benzo [c] chromenes as non-steroidal progesterone receptor antagonists for use against cancers. Bioorganic & Medicinal Chemistry. 2021;32:116003.
- Kassem N, Araya-Secchi R, Bugge K, Barclay A, Steinocher H, Khondker A, et al. Order and disorder—An integrative structure of the full-length human growth hormone receptor. Science Advances. 2021;7(27):eabh3805.
- Basu R, Nahar K, Kulkarni P, Kerekes O, Sattler M, Hall Z, et al. A novel peptide antagonist of the human growth hormone receptor. Journal of Biological Chemistry. 2021;296.
The author declares that he has no competing interests.
Fund: No fund
Author Contribution: The author did all the related work.
Data and Materials Availability: All data are available in the main text or the supplementary materials.
(ISSN - Online)
2959-8591
