The Internet has become an indispensable part of modern life, providing access to information on an unprecedented scale. However, this digital landscape also presents an increasing number of security risks, including the proliferation of malicious URLs, often hidden within emails, social media posts, and malicious website browsing experiences. When a user accidentally clicks on a malicious URL, it can cause a variety of damage to both the user and the organization. These URLs can redirect users to phishing sites that cybercriminals have carefully designed to look like legitimate sites, such as banks, online retailers, or government agencies. These phishing sites aim to trick users into voluntarily divulging sensitive information, including usernames, passwords, credit card numbers, Social Security numbers, and other important personal data, which can result in serious damage such as financial loss and the use of the data to defraud others (1). The continued development of phishing sites, which often use advanced social engineering techniques, increases the risk of exploiting users’ trust despite their security awareness training (2). Malicious URLs are one of the most common ways malware spreads. A single click on a malicious URL can trigger the download a installation of a wide range of malware, including viruses, Trojans, ransomware, spyware, and keyloggers without the user noticing (3). These malicious programs can compromise the user’s device, steal data, encrypt files and demand a ransom to decrypt them, monitor user activity, or even give attackers complete remote control over the device on which the malware is installed. Ultimately, this can cause financial, operational, or reputational damage to companies and organizations that hold user data (4). Traditional methods, such as blacklisting, fail to effectively identify newly emerging threats to detect malicious URLs, as these methods rely on pre-defined malicious URLs, leaving a gap in protection against unknown or newly created malicious links. Attackers are constantly working to circumvent blacklists by constantly creating new URLs and using techniques such as URL shortening and domain spoofing (creating domains that visually resemble legitimate ones) (5). Furthermore, attackers use sophisticated social engineering techniques, crafting convincing messages and deceptive links that exploit human psychology to lure users into clicking and effectively bypass many technical defenses (6). Whitelisting, an alternative approach to blacklisting where only pre-approved URLs are allowed, severely restricts user access and is often impractical for general Internet use. Machine learning has emerged as a powerful tool in the field of cybersecurity, providing more dynamic and efficient solutions to address these sophisticated threats by harnessing the power of data analysis. Machine learning algorithms can learn patterns and characteristics associated with malicious URLs, enabling them to accurately classify unknown URLs (7). Unlike traditional systems, machine learning models can adapt to new URL patterns and identify previously unseen threats, making them a critical component of proactive cybersecurity protection. Deep learning enhances this capability further by detecting subtle indicators of maliciousness (8) that traditional methods or even simple machine learning approaches may miss. Despite the accuracy that deep learning models may provide, they require more time in the detection process, prompting us to consider a way to combine the speed of traditional machine learning models with the accuracy of deep learning models (9,10). This leads us to explore ensemble models. This research focuses on exploring the effectiveness of machine learning and deep learning techniques for detecting malicious URLs, specifically investigating the potential of ensemble learning methods to enhance the accuracy and efficiency of detection. We aim to contribute to the advancement of cybersecurity by:
- Analyzing the essential components and features of URLs: Extracting the essential lexical features that distinguish benign from malicious URLs. This will include a deep dive into the structural elements of URLs and an exploration of how features such as URL length, character distribution, presence of specific keywords, and domain characteristics can be used to identify potentially malicious URLs.
- Investigating the performance of various classification algorithms: Discovering the most efficient models for URL classification. This will include a comparative analysis of different machine learning algorithms, including both traditional methods (e.g., support vector machines and naive Bayes) and more advanced deep learning methods (e.g., convolutional neural networks and recurrent neural networks). The goal is to identify the algorithms that are best suited to the specific task of detecting malicious URLs, taking into account factors such as accuracy and speed.
- Proposing and testing ensemble learning techniques: Exploring the benefits of combining multiple models to improve accuracy and reduce training time. Ensemble techniques such as bagging and stacking offer the potential to leverage the strengths of different individual models, creating a more robust and accurate detection method overall.
This research specifically investigates the effectiveness of clustering techniques, especially bagging and stacking, in the context of detecting malicious URLs. First, we extract and analyze lexical features from the dataset, pre-process the data, and then compare the performance of several classification algorithms, including traditional machine learning models, deep learning, and ensemble learning. Finally, we evaluate the effectiveness of bagging and stacking techniques, highlighting their potential to enhance detection capabilities and reduce training and testing time, thus enhancing cybersecurity measures against malicious URL threats. Detecting malicious URLs has been an important focus of cybersecurity research, with many studies exploring a wide range of machine learning, deep learning, and ensemble methods. These efforts can be categorized based on the approach used to detect malicious URLs.
Machine Learning Classifiers
The basic approach involves applying traditional machine learning classifiers. Xuan et al. (11) investigated the use of support vector machines (SVM) and random forests (RF) to distinguish malicious URLs. Their dataset included 470,000 URLs, using an imbalanced dataset (400,000 benign and 70,000 malicious). While the random forest showed superior predictive effectiveness, the training time was quite long. However, the testing time was similar. Vardhan et al. (12) performed a comparative analysis of several supervised machine learning algorithms. These included naive Bayes, k-nearest neighbors (KNN), stochastic gradient descent, logistic regression, decision trees, and random forest. They used a dataset of 450,000 URLs obtained from Kaggle. Of these, the random forest consistently achieved the highest accuracy. However, a major limitation identified was the high computational cost associated with the random forest, which hinders its deployment in real-time applications. Awodiji (13) focused his research on mitigating threats such as malware, phishing, and spam by applying SVM, naive Bayes, decision trees, and random forests. For training and evaluation, he used the ISCX-URL-2016 dataset from the University of New Brunswick, known for its diverse representation of malicious URL types. The random forest algorithm achieved the best accuracy (98.8%), outperforming the other algorithms. However, the study lacks specific details regarding the training time and computational resource requirements of each algorithm, making it difficult to evaluate their overall efficiency. Velpula (14) proposed a random forest-based machine learning model that combined lexical, host-based, and content-based features. This approach leveraged a dataset from the University of California, Irvine, Machine Learning Repository containing 11,000 phishing URLs. The dataset was rich in features, including static features (e.g., domain age and URL length) and dynamic features (e.g., number of exemplars and external links). While the combination of diverse features significantly improved the model’s accuracy to 97%, the research did not explore the potential of other machine learning algorithms. Reyes-Dorta et al. (15) explored the relatively new field of quantum machine learning (QML) for detecting malicious URLs and compared its effectiveness with classical machine learning techniques. They used the “Hidden Phishing URL Dataset,” which included 185,180 URLs. Their results showed that traditional machine learning methods, especially SVM with Radial Basis Function (RBF) kernel, achieved high accuracy levels (above 90%). The research also highlighted the effectiveness of neural networks but noted that the current limitations of quantum hardware hinder the widespread application of QML in this field, making traditional machine learning models perform better due to their continuous improvement.
Deep Learning Models
Deep learning, with its ability to learn complex patterns from data, has emerged as a promising approach for detecting malicious URLs. Johnson et al. (16) conducted a comparative study of traditional machine learning algorithms (RF, C4.5, KNN) and deep learning models (GRU, LSTM, CNN). Their study confirmed the importance of lexical features for detecting malicious URLs, using the ISCX-URL-2016 dataset. The results indicated that the GRU (Gated Recurrent Unit) deep learning model outperformed the Random Forest algorithm. However, the researchers did not compare them with other machine learning and deep learning algorithms to explore whether they achieve better accuracy. Aljabri et al. (17) evaluated the performance of both machine learning models (Naive Bayes, Random Forest) and deep learning models (CNN, LSTM) in the context of detecting malicious URLs. The researchers used a large, imbalanced dataset obtained by web crawling with Mal Crawler. 1.2 million URLs were used for training, of which 27,253 were considered malicious, 1,172,747 were considered benign, and 0.364 million URLs were used for testing. The dataset was validated using Google’s Safe Browsing API. The results showed that the Naive Bayes model achieved the highest accuracy (96%). However, the study had limitations, including unexplored potential of other machine learning and deep learning algorithms, and uneven distribution within the dataset. These limitations may limit the generalizability of the results and potentially introduce bias into the model. Gopali et al. (18) proposed a new approach by treating URLs as sequences of symbols, enabling the application of deep learning algorithms designed for sequence processing, such as TCN (Temporal Convolutional Network), LSTM (Long Short-Term Memory), BILSTM (Bidirectional LSTM), and multi-head attention. The study specifically emphasized the important role of contextual features within URLs for effective phishing detection. Their results confirmed that the proposed deep learning models, particularly BILSTM and multi-head attention, were more accurate than other methods such as random forests. However, the study used a specialized dataset, limiting the generalizability of the results to other URL datasets, and did not comprehensively evaluate a broader range of other deep learning and machine learning algorithms.
Ensemble Learning Approaches
In addition to single classifiers, ensemble approaches, which combine multiple models, have been explored to improve detection performance. Chen et al. (19) leveraged the XGBoost algorithm, a boosting algorithm. Boosting is a popular ensemble learning technique known for its classification speed. Their work emphasized the importance of lexical features in identifying malicious URLs. Through feature selection, they initially identified 17 potentially important features, and then refined them to the nine best features to reduce model complexity while maintaining a high accuracy of 99.93%. However, the study did not provide a sufficiently detailed description of the required training time and computational resources consumed by the XGBoost model.
Feature Engineering and Selection
Recognizing the importance of feature quality to model performance, some research has focused specifically on feature engineering and selection techniques. Oshingbesan et al. (20) sought to improve malicious URL recognition by applying machine learning with a strong focus on feature engineering. Their approach involved the use of 78 lexical features, including hostname length, top-level domain, and the number of paths in a URL. Furthermore, they introduced new features called “benign score” and “malicious score,” derived using linguistic modeling techniques. The study evaluated ten different machine learning and deep learning models: KNN, random forest, decision trees, logistic regression, support vector machines (SVM), linear support vector machines (SVM), feed-forward neural networks (FFNN), naive Bayes, K-Means, and Gaussian mixture models (GMM). Although the K-Nearest Neighbor (KNN) algorithm achieved the highest accuracy, it suffers from significant drawbacks in terms of training and prediction time requirements. Mat Rani et al. (21) emphasized the critical role of selecting effective features for classifying malicious URLs. They used information acquisition and tree-shape techniques to improve the performance of machine learning models, particularly in the context of phishing site detection. The study used three classifiers: Naive Bayes, Random Forest, and XGBoost. Features selected using the tree-shape technique showed a significant positive impact on accuracy. While XGBoost achieved the highest accuracy of 98.59%, the study did not fully explore the potential of other deep learning algorithms or delve into aspects of model efficiency, such as their speed and resource requirements during the training and testing phases. Even though machine learning and deep learning methods have achieved high accuracy in identifying malicious URLs, there are concerns regarding training and prediction time efficiency and the complexity of tuning hyperparameters (11–21). Ensemble methods, such as Random Forest and XGBoost, are effective due to their ability to handle high-dimensional data, improve accuracy, and reduce the overfitting problem. However, they often require higher computational requirements (20), (21). Despite the great efforts made by researchers to detect malicious URLs, critical analysis reveals several points that need to be explored and require further attention.
- Real-Time Applications: Most studies have focused on achieving high accuracy but do not delve into time efficiency, which is critical for detecting malicious URLs, especially in light of the rapid technological development. This limitation raises concerns about the feasibility of using these models in real-time applications (11–21).
- Data Imbalance: Most research on datasets suffers from an imbalance between benign and malicious URLs (11–21). This imbalance significantly impacts model training and may bias the model’s performance in favor of the dominant class. Techniques such as over-sampling and under-sampling are needed to address this issue for more reliable evaluation.
- Feature Extraction and Selection: Some research shows the need to explore how features are extracted, transformed, and selected effectively to improve training and prediction, efficiency, and accuracy(14,17,19–21).
MATERIALS AND METHODS
Hardware Specifications
The experiments in this research were conducted using Google Colaboratory (Colab) with virtual CPU settings to ensure methodological consistency. Colab operates on a dynamic resource allocation model, and the predominant configuration consists of an Intel Xeon processor with two virtual central processing units (vCPUs) and 13 GB of RAM. Acknowledging that there is potential for slight inter-session variations in resource assignment.
Dataset
The dataset (benign and malicious URLs) (22) used in this research consists of 632,508 rows with an equal distribution of 316,254 benign URLs and 316,254 malicious URLs, categorized according to the three columns, “url”, “label”, and “result”, which contain the URL itself, the corresponding classification label (either ‘Benign’ or ‘Malicious’), and the classification result as an integer value (0 for benign and 1 for malicious), We extracted a total of 27 lexical features from each URL as shown in Table 2.
Data Preprocessing
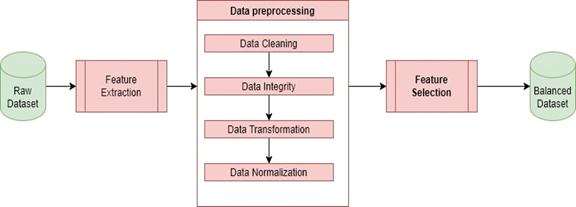
Data preprocessing is critical to achieving reliable and accurate results, as missing values and inconsistencies can introduce significant bias during the training process, leading to inaccurate predictions. Preprocessing steps,such as cleaning, integration, transformation, and normalization, improve model performance and prevent overfitting by ensuring data consistency and representation.
Data Cleaning
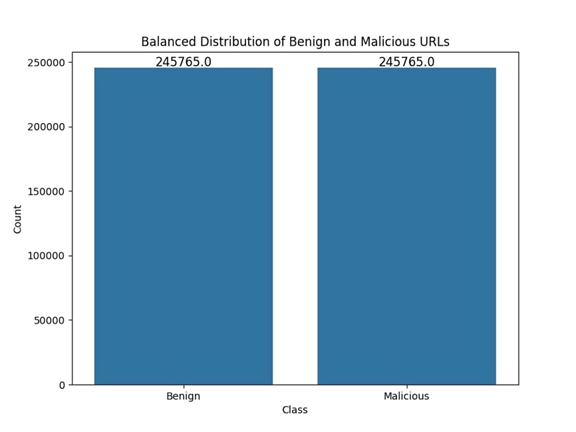
The missing values (NAN) and inconsistent data within the dataset are removed, ensuring its completeness and accuracy to train the model reliably. After the deletion process, the dataset became unbalanced. To overcome this problem and rebalance the dataset, the Random Under Sampling technique was used, where samples from the majority class were randomly deleted. The resulting balanced dataset was saved to complete other pre-processing steps on it. Figure 1 shows the balanced distribution of samples:
Data Integrity
Maintaining a consistent data structure by standardizing column names and data formats requires ensuring the dataset does not contain duplicates or inconsistencies in the format of different attributes.
Data Transformation
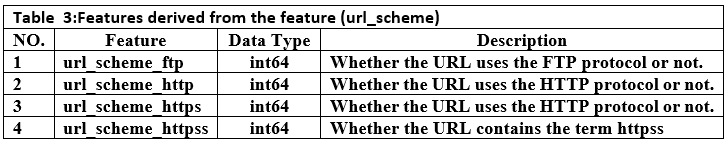
Converting categorical features such as url_scheme and get_tld into a numeric format, which can be easily processed by various machine learning algorithms. This involved converting categorical variables into multiple numeric variables. The url_scheme feature was converted into four features, each representing a single protocol. We got four new features as shown in Table 3. The top-level domain feature (get_tld), which is a categorical feature, was converted using an ordinal encoder to be processed by the algorithms(23).
Data Normalization
Normalizing the data’s numeric attributes was conducted using appropriate techniques. Features such as url_length, path_length, and host length were normalized to achieve better model performance by equalizing the impact of these attributes, which differ in magnitude.
Feature Selection
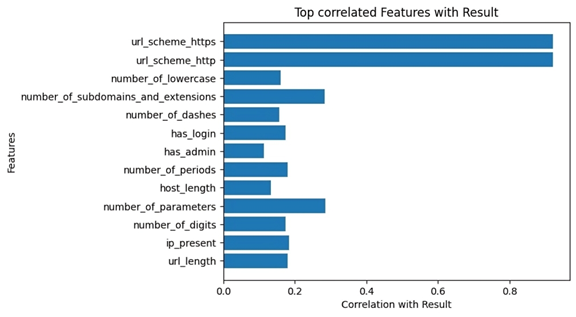
Correlation-based feature selection was used to examine the relationship between features and the target variable. This method is characterized by its rapid feature selection while maintaining classification accuracy. The most influential features that had significant correlations with the target variable and small correlations between them were then selected to reduce redundancy and simplify model building (24,25). After selecting the features that were most correlated with the target variable [‘result’], thirteen independent variables (features) were selected, as shown in Figure 2. Figure 3 shows the data preparation process, illustrating all the steps taken to obtain a balanced dataset.
Proposed Solution
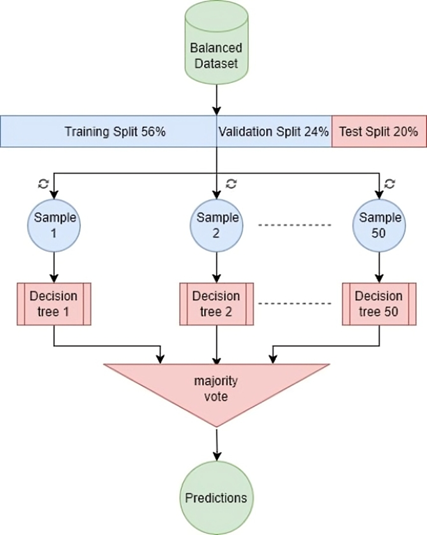
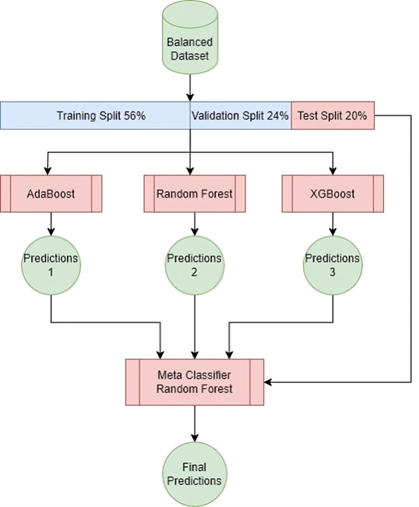
This study proposes an innovative approach to detecting malicious URLs using ensemble learning techniques, specifically Bagging (Bootstrap aggregation) and stacking. Bagging (Bootstrap aggregation) uses 50 decision trees as its baseline models, yielding better results than using more or fewer trees, as shown in Tables 9 and 10. Majority voting is used to obtain the final predictions, as shown in Figure 4. While stacking uses models (AdaBoost, Random Forest, and XGBoost) as base models and uses a random forest as meta model to obtain the final predictions, as shown in Figure 5. These techniques combine predictions from multiple base learners, resulting in a faster and more accurate classification model. Bagging is a statistical procedure that creates multiple datasets by sampling the data with replacement to obtain a final prediction result with minimal variance(26). Stacking combines weak learners to create a strong learner. It combines heterogeneous parallel models to reduce bias in these models. Stacking is also known as stacked generalization. Similar to averaging, all models (weak learners) contribute based on their weights and performance, to build a new model on top of the others(27). Models (AdaBoost, Random Forest, and XGBoost) were used as weak learners to gain different perspectives on the dataset and avoid duplicate predictions.
Verifying the Results
To analyze the effectiveness of the proposed solution extensively, a comparison of its performance with many traditional machine learning algorithms and deep learning techniques is applied. The algorithms that were implemented and evaluated include:
Traditional Machine Learning Algorithms
The machine learning algorithms evaluated included several with specific parameter settings. The Decision Tree was configured with random_state=42 for consistent results. Logistic Regression used max_iter=5000 and random_state=42. The SVM was trained with a linear kernel (kernel=’linear’) and random_state=42. Finally, K-Nearest Neighbors was set to consider 3 neighbors (n_neighbors=3). Gaussian Naive Bayes and Bernoulli Naive Bayes were used with default parameter settings without adjustments.
Deep Learning Algorithms
The deep learning models, CNN, FFNN and RNN are set using various parameters to adjust the model’s performance. The CNN has several convolutional layers and max-pooling layers, a flatten layer, and two Dense layers. The FFNN had set Adam as the model optimizer, has an initial learning rate of 0.001, each layer having different number of parameters. The FFNN had three Dense Layers. RNN has two Simple RNN layers and also uses Adam. Finally, the Radial Basis Function Network has set hidden_layer_sizes= (10), the maximum iterations are set to 1000 iterations.
Ensemble Learning Algorithms
The ensemble learning algorithms employed a variety of configurations to create robust predictive models. The initial Voting Classifier was set to use a hard voting strategy. The initial Stacking Classifier integrated a Decision Tree Classifier with random_state=42 as its final estimator, utilized all available cores (n_jobs=-1), and employed passthrough. Bagging Classifiers were configured with 50 base estimators (n_estimators=50), a Decision Tree Classifier (with the default random state) as the base estimator, a max_samples value of 0.80, specified bootstrap sampling, and a random_state of 42. AdaBoost used 100 estimators (n_estimators=100), a Decision Tree Classifier with max_depth=10 as the base estimator, a learning rate of 0.5, and random_state=42. The final Voting and Stacking classifiers were then set up in the same way. Gradient Boosting and Extra Trees utilized a fixed random_state.
Model Evaluation
To evaluate model performance, we employed a comprehensive set of metrics, including:
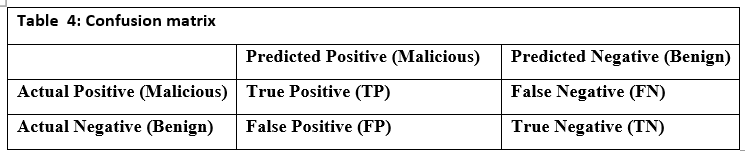
- Confusion Matrix: Table 4 shows the confusion matrix, which compares predicted classifications to actual labels, revealing True Positives (TP), True Negatives (TN), False Positives (FP), and False Negatives (FN). This is crucial for binary classification tasks.
- Accuracy: Overall correct predictions.
![]()
Precision: Correctly predicted malicious URLs out of all those predicted as malicious, where high precision minimizes false positives.
![]()
- Recall: Correctly identified malicious URLs out of all actual malicious URLs, where high recall minimizes false negatives.
![]()
- Specificity: Correctly identified benign URLs out of all actual benign URLs.
![]()
- F1 Score: Harmonic mean of precision and recall.
![]()
Training and Prediction Time
- Training Time: Training time measures the time taken to train each model on the training data, providing insights into the efficiency and scalability of different learning algorithms.
- Prediction Time: Prediction time quantifies the time required for each trained model to predict the classification of a new URL and assesses the model’s suitability for online URL filtering applications that require fast responses to incoming URLs, impacting the model’s applicability in real-time systems. In this paper, we calculated the above metrics for each of the algorithms considered in our research, resulting in a comparative performance analysis that reported on the selection of the optimal model.
RESULT
This section presents the results obtained from the implemented algorithms, discusses their performance, and compares their strengths and limitations. To evaluate the models, we focus on accuracy, precision, recall, specificity, F1 score, training time, and prediction time for each model to provide a comprehensive analysis of their effectiveness in detecting malicious URLs.
Individual Machine Learning Models
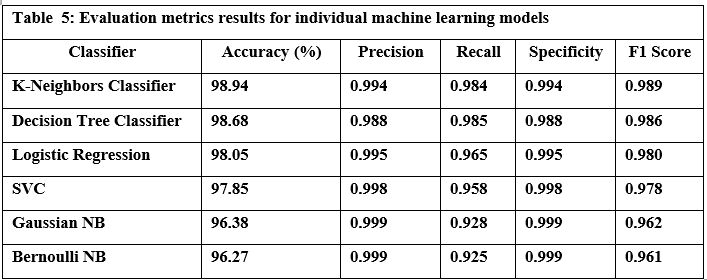
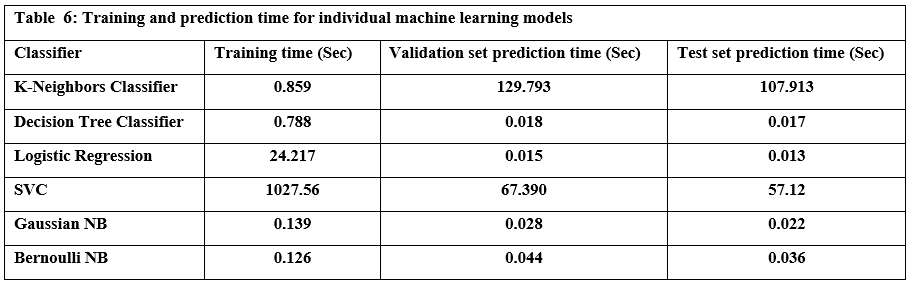
Tables 5 and 6 summarize the performance of six common machine learning algorithms, namely K-Nearest Neighbors (KNN), Decision Tree, Logistic Regression, Support Vector Classifier (SVC), Gaussian Naive Bayes, and Bernoulli Naive Bayes, evaluated based on several key metrics to provide a clear picture of their performance in classifying URLs as benign or malicious. Based on the results of practical experiments on individual machine-learning models, we summarize the following:
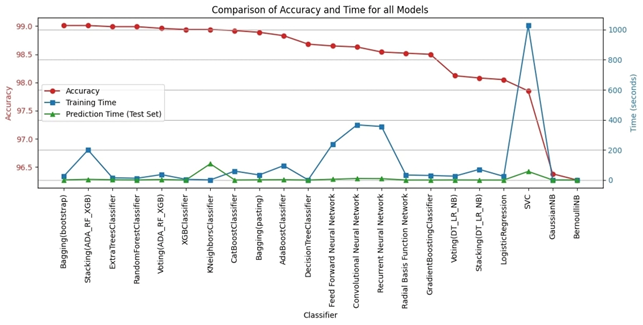
The K-Nearest Neighbors (KNN) model achieved the highest accuracy, reaching 98.94%, while the Bernoulli Naive Bayes (Bernoulli NB) model exhibited the lowest accuracy at 96.27%. Drilling down into individual metrics, Bernoulli NB demonstrated exceptional precision of 0.999, effectively identifying benign URLs. However, the Decision Tree model excelled in recall 0.985, successfully identifying malicious URLs. Bernoulli NB also showed the best specificity. Finally, KNN displayed the best-balanced performance, as measured by the F1 score, which considers both precision and recall. The models also varied significantly in terms of speed. Bernoulli NB was the quickest in training at 0.126 seconds, whereas the Support Vector Machine (SVC) model required substantially more time 17 minutes, possibly due to the size of the dataset used for training and model optimization. For prediction, Logistic Regression outperformed all others, whereas KNN had the longest prediction times. These results illustrate a crucial tradeoff between computational efficiency and predictive power, where simple and easily trained models require less computational overhead, whilst algorithms that model complex non-linear patterns typically require a considerably greater level of computing time.
Deep Learning Models
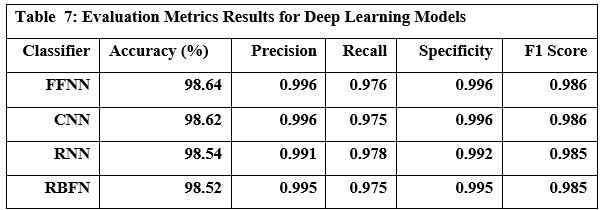
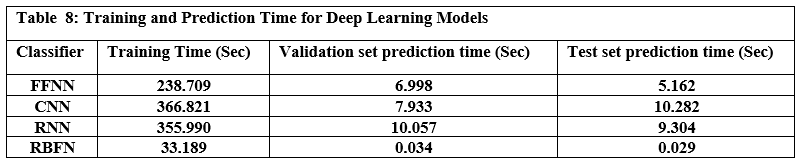
Table 7 presents the performance metrics for four prominent deep learning models FFNN (Feed Forward Neural Network), CNN (Convolutional Neural Network), RNN (Recurrent Neural Network), and RBFN (Radial Basis Function Network), while Table 8 shows the time each model took to train and predict. Based on the results of practical experiments on deep learning models, we concluded the following: The Feed-Forward Neural Network (FFNN) achieved the highest accuracy at 98.64%, while the Radial Basis Function Network (RBFN) had the lowest accuracy at 98.52%. While the accuracy differences were small, other metrics showed some variation; the Convolutional Neural Network (CNN) showed the highest precision and specificity, indicating its ability to correctly identify both benign and malicious URLs, whereas the Recurrent Neural Network (RNN) achieved the highest recall, showing high effectiveness in capturing actual malicious URLs, despite it only showing the second highest accuracy level. Ultimately, the FFNN model exhibited the highest F1 score. Regarding speed, the RBFN model proved to be the most computationally efficient in terms of training time, completing the training process in 33.189 seconds, compared to the CNN, which required over 10 times longer. It is noteworthy to remember the significantly increased computational power required for the CNN. Furthermore, RBFN was also the fastest model for making predictions.
Ensemble Learning Models
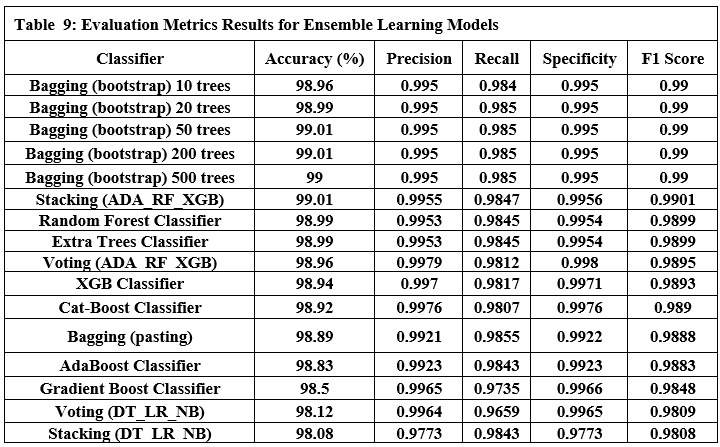
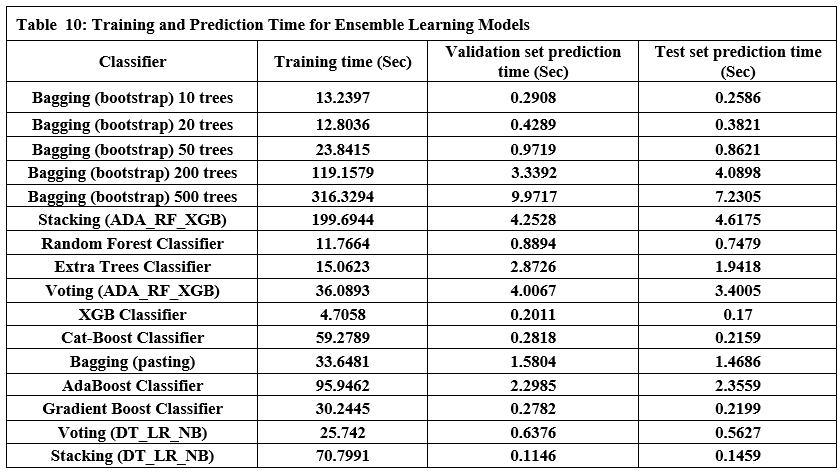
Tables 9 and 10 show the performance of twelve ensemble learning models using their three techniques (bagging, stacking, and boosting). Regarding experiments on ensemble learning models, we note the following:
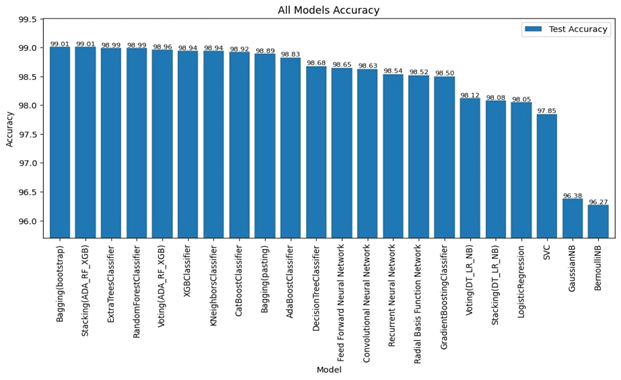
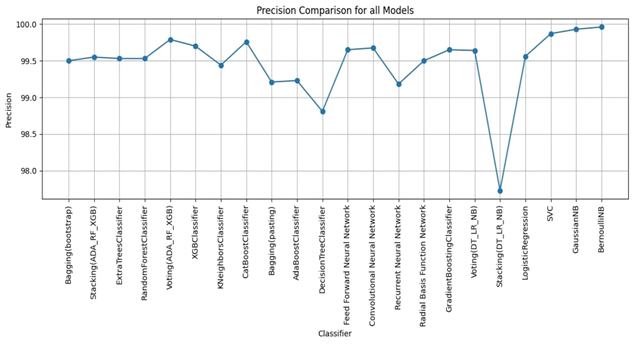
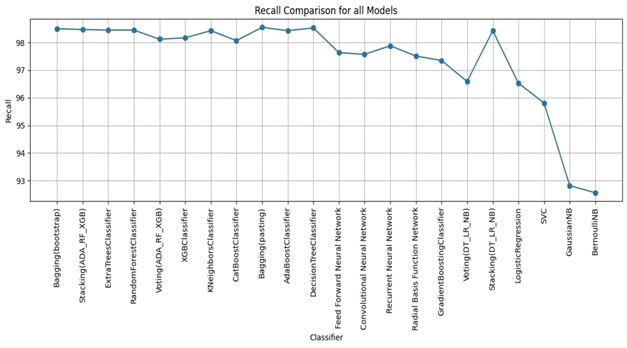
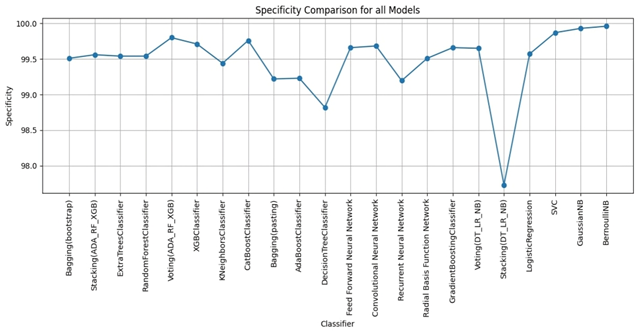
Bagging (bootstrap) is the top performer in terms of accuracy, reaching 99.01%. At the other end of the spectrum, the stacking model combining Decision Trees, Logistic Regression, and Naive Bayes showed the lowest accuracy. Examining other key metrics, the Voting model incorporating Adaboost, Random Forest, and XGBoost models achieved both the highest precision and specificity. Interestingly, Bagging (pasting), a variation of the Bagging algorithm, demonstrated the highest recall. For the best-balanced performance, reflected in the F1-score, stacking combining Adaboost, Random Forest, and XGBoost produced the highest F1-score. The speed varied substantially across the different ensemble techniques investigated. In the training process, the individual XGBoost model was significantly faster. In contrast, the Stacking model incorporating Adaboost, Random Forest, and XGBoost, was by far the slowest to train. For prediction speed, the stacking model (Decision Trees, Logistic Regression, Naive Bayes) demonstrated speed during prediction. The slowest prediction time, unsurprisingly, was seen with Stacking (Adaboost, Random Forest, and XGBoost), confirming that the complexity incurred through higher-level models impacts both training and testing times within the model. A comprehensive performance evaluation of all models highlights notable differences in strengths and weaknesses. Figure 6 provides a comparison of the overall accuracy achieved by each model, while Figures 7, 8, 9, and 10 visualize other critical metrics of model evaluation: precision, recall, specificity, and F1 score, respectively.
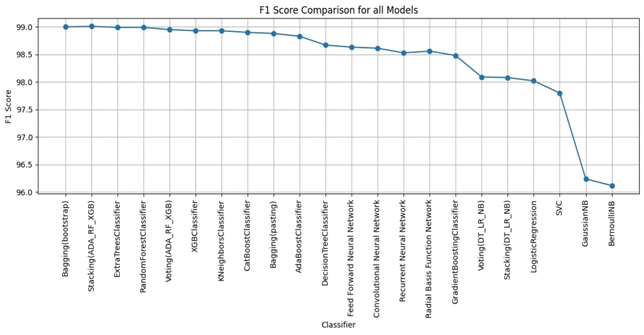
DISCUSSION
These findings demonstrate a significant correlation between the characteristics of URLs and their likelihood of being designated as malicious, underscoring the necessity of precise feature extraction for the efficacious identification of malevolent URLs. Furthermore, using well-preprocessed datasets leads to accurate classification results. Moreover, the precision and efficiency of the model in terms of classification or prediction are contingent upon the type and integrity of the data utilized. The selection of an appropriate model pertinent to the specific issue at hand is of paramount importance, as the correct model selection fosters accurate classifications and predictions at a high rate, resulting in the development of a reliable classifier. The results for traditional Machine Learning algorithms showed moderate accuracy. Most Machine Learning models, such as Logistic Regression and Naive Bayes, underperformed the proposed Ensemble models. This may be attributed to limitations such as overfitting or feature dependency in individual Machine Learning algorithms. The high accuracy achieved by deep learning models stems from their ability to handle intricate relationships within the data, although the computation costs involved with training these complex models can be considerable. However, Ensemble Learning techniques consistently outperformed both individual Machine Learning and Deep Learning techniques. In particular, bagging with bootstrap sampling (Bagging (Bootstrap)) consistently exhibited exceptional accuracy while minimizing training and prediction times. The highest accuracy achieved was with Bagging (Bootstrap), which obtained 99.01%. This suggests that Bagging is the optimal approach for a real-time, practical system for malicious URL detection. Stacking demonstrated similar performance levels with slightly extended training durations due to its reliance on a structure consisting of several models. The proposed solution resulted in the following benefits of ensemble learning: Improved accuracy: By combining multiple models, ensemble learning often achieves significantly higher accuracy than individual learners. This is because each model learns from different aspects of the data, thus reducing bias and variance. Several studies highlight the advantage of ensemble methods, including “Bagging Predictors” by Breiman (28), which shows significant improvement in accuracy and reduced variance compared to individual learners. Improved generalization: Ensemble learning often produces more robust models with improved generalization to unseen data, which helps mitigate overfitting. The article “Stacked Generalization” by Wolpert (27) improved the generalization ability of ensemble techniques, leading to better model performance on unseen data. Robustness to Noise and Outliers: Ensemble learning tends to be less sensitive to noise and outliers in the data, which increases model stability. The paper on “XGBoost: A scalable tree boosting system” by Chen and Guestrin (29) emphasizes XGBoost’s robust handling of noise and outliers, which contributes to overall model stability. Increased Stability: By calculating average predictions from multiple models, ensemble learning generally produces more consistent results than individual models, reducing variability in performance. Work on “Bagging Predictors” by Breiman (28) highlights how Bagging improves consistency by combining multiple predictions, reducing variability, and making models more stable. Reduce Complexity: While ensemble models may seem complex, they may sometimes simplify the learning process, especially when compared to complex deep learning, providing a better balance between accuracy and complexity. Some studies, such as “highly random trees” by Geurts et al. (30), have noted this advantage. Overall, these results, shown in Figure 11, strongly support the hypothesis that ensemble learning, and in particular Bagging (Bootstrap), is an effective technique for accurately detecting malicious URLs. It surpasses traditional machine learning algorithms in accuracy and performance, and demonstrates more favorable trade-offs between accuracy, computational complexity, and speed when compared to Deep Learning models.
CONCLUSION AND RECOMMENDATION
This study conducted a systematic evaluation of a range of machine learning, deep learning, and ensemble learning techniques for the purpose of detecting malicious URLs. Feature selection was employed, prioritizing those exhibiting the strongest correlation with the dependent variable, resulting in the selection of 13 lexical features from a total of 27 extracted from the dataset. The results demonstrate the superior performance of ensemble learning methods, specifically the Bagging (Bootstrap) technique, in achieving high accuracy alongside rapid training and prediction capabilities. This approach surpassed the accuracy of individual models and the speed of deep learning models, underscoring its effectiveness in mitigating the growing cybersecurity threat posed by malicious URLs. The speed and accuracy of the Bagging (Bootstrap) make it very useful for cybersecurity. It could be a strong tool in real-time systems for detecting and blocking threats.

![Determination of copper Ions (II) in Aqueous Solutions Using Organic Reagent 3-Hydroxy-4-[(2-hydroxy benzylidene) amino] naphthalene Sulphonic Acid-1 (HHBAN) by Spectrophotometric method](https://journal.hcsr.gov.sy/wp-content/uploads/2025/07/صورة1.jpg)