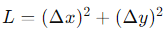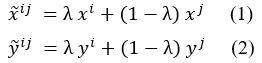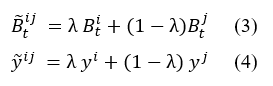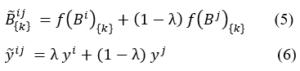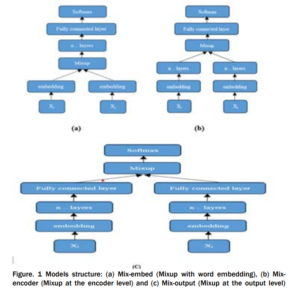
INTRODUCTION
Vascular Endothelial Growth Factor (VEGF) is a homodimeric glycoprotein with a molecular weight of approximately 28 KD. This protein serves as a critical mediator for angiogenesis, the formation of new blood vessels. VEGF exerts its physiological effects by binding to two receptors known as Flt-1 (VEGFR-I) and KDR (VEGFR-II) (1). These receptors possess seven extracellular immunoglobulin-like domains and one intracellular tyrosine kinase domain. They are expressed on the surface of endothelial cells, playing a critical role in the regulation of angiogenesis (2, 3). Under hypoxic conditions, the expression of VEGF receptors increases, leading to the induction of the angiogenesis phenomenon. The binding of VEGF to the VEGFR-II receptor stimulates the response of endothelial cells in blood vessels (4-6). Although VEGFR-I has a higher affinity for this factor, it only plays a role in sequestering VEGF and facilitates its access to the VEGFR-II receptor, which is the primary receptor mediating antigenic signaling. VEGF binds to these receptors through two distinct domains (7, 8). In healthy individuals, VEGF contributes to angiogenesis during embryonic development and plays a critical role in adult wound healing. Pathological angiogenesis is pivotal in various conditions such as tumor growth, metastasis, diabetic retinopathy, macular degeneration, rheumatoid arthritis, and psoriasis (9-11). Inhibition of angiogenesis can be achieved by several methods, including inhibition of endothelial cell signal transduction, migration, and survival, as well as modulation of factors such as growth, proliferation, matrix metalloproteinase, and bone marrow precursor cells. VEGF, as a regulator of tumor angiogenesis, exerts its angiogenic effects by binding to its receptors, especially VEGFR-II, thereby influencing the mentioned processes (12-14). Therefore, this molecule is of great importance as a valuable drug target in antiangiogenic therapies (12, 15). Single-domain antibodies, nanobodies, or VHHs possess valuable characteristics, including effective tissue penetration, high stability, and ease of humanization, efficient expression in prokaryotic hosts, and specificity and affinity for their respective antigens. Consequently, they can be introduced as alternative therapeutic candidates to traditional antibodies. VHHs represent the smallest functional unit of an antibody, preserving all of its functions, and due to their minimal size, they are also recognized as nanobodies (16-18). The studies conducted by Shahngahian and colleagues in 2015 demonstrated that VEvhh10 (accession code LC010469) exhibits a potent inhibitory effect on the binding of VEGF to its receptor (19). The mentioned VHH exerts its inhibitory role by binding to the VEGF receptor binding site. VEvhh10 also possesses the highest binding energy at the VEGF receptor binding site among other members of the VHH phage display library, covering vital amino acids involved in the biological activity of VEGF and disrupting its biological function. A standard method for expressing non-fused VHH is the use of E. coli expression systems. However, expressing non-fused VHH using conventional cytoplasmic expression methods in this prokaryotic host often leads to the formation of inclusion bodies (20, 21). In this study, to overcome this issue, a surface display technique was chosen, allowing peptides to be presented on the surface of microbial cells through fusion with anchoring motifs. The ice nucleation protein (INP) is one of the joint membrane proteins in bacteria (22-27). Despite the size limitations that most surface expression systems have regarding the display of target proteins on the cell surface, an INP-based surface expression system can express and display VHH on the bacterial surface, and overcoming the problems of cytoplasmic expression within bacteria.
MATERIALS AND METHODS
Molecular and Chemical Materials
The materials, chemicals, and reagents required for the lab are listed as follows:
- Ampicillin and Agarose from Acros (Taiwan)
- IPTG from SinaClon (Iran)
- Ni-NTA resin from Qiagen (Netherlands)
- Plasmid extraction kit and gel extraction kit from GeneAll (South Korea)
- Enzyme purification kit from Yektatajhiz (Iran)
- Restriction enzymes HindIII/XhoI, ligase enzyme, and other molecular enzymes from Fermentas (USA).
- pfu polymerase and Taq polymerase from Vivantis (South Korea)
- Primers from Sinagen (Iran)
- Methylthiazole-tetrazolium (MTT) powder from Sigma (USA)
- Penicillin-Streptomycin, Trypsin-EDTA, and DMED-low glucose from Bio-Idea (Iran)
- Fetal Bovine Serum (FBS) from GibcoBRL (USA)
- A monoclonal conjugated anti-human antibody with HRP from Pishgaman Teb (Iran)
- Anti-Austen antibody from Roche (Switzerland)
- Other chemicals from Merck (Germany)
Design of fusion genes and constructions of pET-INP-VHH
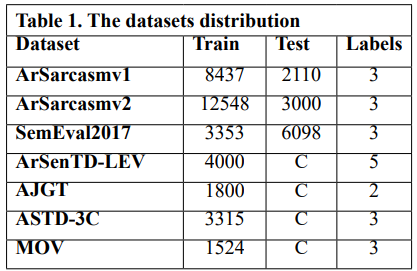
Gene Cloning: Primers for the amplification of the VEvhh10 gene with the accession code LC010469 were initially designed (Table 1). The plasmid containing the gene fragment served as a template for the Polymerase Chain Reaction (PCR). Amplification was carried out using Taq polymerase and Pfu polymerase enzymes in a thermocycler with a programmed temperature profile (Table 2). Various annealing temperatures for primer binding to the template were tested, and a temperature of 65°C was found to be the optimal annealing temperature. Primer cutting sites at the beginning and end of the Gene were designed, and the location of the TEV protease enzyme cutting site was positioned between the INP linker and the VEvhh10 sequence.
The VEvhh10 gene fragment, amplified by the pfu polymerase through PCR, was extracted from the gel. Simultaneously, digestion of this fragment and the pET-21a vector containing the INP linker was carried out using the HindIII and XhoI restriction enzymes. Purification was performed using a commercial enzyme purification kit. Ligation of the gene fragment into the target vector was accomplished using the T4 DNA ligase enzyme. The resulting product was incubated at 4°C overnight. The ligated product was then transferred into E. coli DH-5α bacteria. To confirm the Gene insertion into the vector, the transformed bacteria were plated on antibiotic- ampicillin containing plates. The obtained colonies were then subjected to Colony PCR using specific primers for VHH, T7 promoter, and terminator primers. The PCR products were analyzed on a 1% agarose gel, and positive transformants were screened and cultured. The recombinant plasmid was purified using a plasmid extraction kit. Enzymatic dual digestion by HindIII and XhoI was performed to confirm the insertion of the gene fragment into the plasmid.
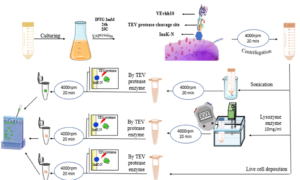
Expression and detection of the Gene constructs InaK-N_TEV Protease
The gene structure of the InaK-N_TEV Protease was transformed in the pET-21a plasmid containing INP and VHH using a heat shock method in the E. coli BL21 (DE3) host. For protein expression, a colony from bacteria harboring the recombinant plasmid was inoculated into 10 mL LB medium supplemented with 100 mg/ml ampicillin and incubated at 37 °C with adequate aeration (250 rpm). Subsequently, 1 mL of the grown bacteria was transferred to 50 mL LB medium containing ampicillin and incubated at 37 °C until the OD600 reached approximately 0.6. Finally, expression was induced with 1 mM IPTG for 24 hours at 25 °C. The resulting product was centrifuged at 4000 rpm for 15 minutes, and the obtained pellet was stored at -20 °C until further analysis. To confirm protein expression, SDS-PAGE (Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis) was performed using a 12% acrylamide gel under non-reducing conditions according to the Laemmli method (28).
The expression and purification of VEGF8-109
The pET-28a plasmid containing the VEGF8-109 gene (the domain binding to the VEGF receptor) was transformed into E. coli BL21 (DE3) bacterial cells and cultured at 37 °C overnight in Terrific Broth (TB) medium supplemented with 100 mg/ml kanamycin. Subsequently, induction was carried out with 0.5 mM IPTG at an OD600 of 0.6, and the cells were incubated at 24 °C for 22 hours (29). After centrifugation (5000 g, 15 minutes, 4 °C), the bacterial cells were sonicated in a lysis buffer containing 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, and 1 mM PMSF (pH 8.0). The sonication product was further centrifuged at 12000 rpm for 20 minutes at 4 °C. The resulting supernatant was analyzed using SDS-PAGE for further characterization (30). Protein purification was carried out using affinity chromatography with a nickel column. For this purpose, the protein sample was transferred onto a column that had previously reached equilibrium by washing with a wash buffer (Tris-base (50 mM, pH 8.0) + NaCl (300 mM) + Imidazole (20 mM)). At this stage, proteins lacking a histidine tag were washed out and removed from the column using this buffer. Only the target protein, due to the presence of a histidine tag, remained bound to the column. Subsequently, using the elution buffer (Tris-base (50 mM, pH 8.0) + NaCl (300 mM) + Imidazole (250 mM)) in the presence of a high concentration of imidazole, protein separation was performed. Additionally, purification was conducted using cold buffers to prevent the thermal denaturation of proteins. Following that, dialysis was performed in PBS buffer containing glycerol overnight at 4 °C, and the protein was analyzed using SDS-PAGE. Protein concentration was determined by the Bradford method with BSA as a protein standard (31).
Cell Culture
Human Umbilical Vein Endothelial Cells (HUVEC) were cultured in T25 flasks using Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 5 mM glucose, 2 mM L-glutamine, 10% fetal bovine serum (FBS), 1% penicillin (100 mg/mL), and streptomycin (100 mg/mL). The cells were maintained at 37°C with 5% CO2 and used for subsequent experiments (32).
In vitro endothelial cell proliferation assay
In order to investigate the effect of VEGF8-109 on the growth and proliferation of HUVECs, the MTT colorimetric method was employed (33). A total of 5×103 cells were seeded in each well of a 96-well culture plate. After cell attachment within 24 hours, the culture medium was replaced with fresh medium containing VEGF8-109-RBD protein at various concentrations (10, 30, 60, 120, and 240 ng /mL), and the cells were incubated at 37 °C for 48 hours. Three independent assessments were performed for each treatment. For cell proliferation determination, cells were treated with 0.5 mg/mL MTT for 4 hours at 37 °C, and then the medium was carefully removed. Formazan crystals were dissolved in 100 μL of DMSO, and the absorbance was read at 570 nm using a µQuant plate reader from BioTek (USA).
ELISA-based immunoassay
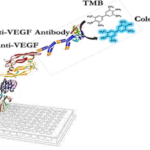
The surface-expressed VHH-expressing bacterial cells, using the INP anchor, were prepared in a carbonate-bicarbonate buffer (pH 9.6, 0.1 M Na2CO3, 0.1 M NaHCO3). Subsequently, 100 μL of bacterial cells containing the INP-VEvhh10 linker were added to each well and incubated at room temperature for 16 hours. After 16 hours, the solution was aspirated, and the wells were washed three times with 100 μL of PBS buffer. Blocking was performed using a blocking buffer consisting of 2% gelatin in PBS (350 μL) for one hour at 37 °C. The blocking solution was then aspirated, and the wells were washed three times with 100 μL of PBST buffer (PBS + 0.05% Tween-20). Serial dilutions of VEGF solutions (ranging from 5.0 ng/mL to 500 ng/mL) were added to the wells and incubated for 2 hours at room temperature. After incubation, all wells were aspirated and washed thoroughly with PBST buffer. Subsequently, 100 μL of human monoclonal anti-VEGF antibody at a concentration of 1000 ng/mL was added to each well, followed by incubation in the dark at 37 °C for 1.5 hours. Then, 100 μL of Anti-Human IgG conjugated with HRP were added to each well and incubated in the dark at 37 °C for 1.5 hours. Finally, 100 μL of TMB and H2O2 were added to each well and incubated for 15 minutes in the dark. The reaction was stopped by adding 100 μL of 2 N sulfuric acid to each well. The absorbance of the wells was then read at 450 nm wavelength.
RESULTS
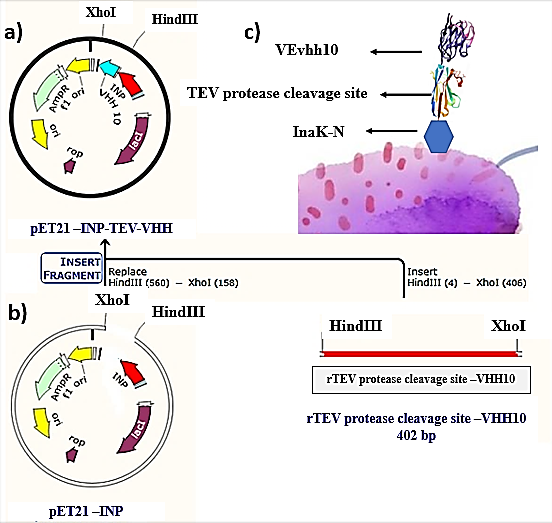
Construction of pET-21a-Ina_K537-TEV-VEvhh10 expression plasmid
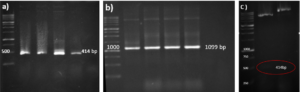
A schematic representation of the gene structure pET-21a-Ina_K537-TEV-VEvhh10 is presented in (Figure 1) using Snapgene.v5.1.5 software. Initially, the plasmid pComb3X containing the VEvhh10 gene sequence with the accession number LC010469 in the GenBank database was registered and extracted from E. coli TG bacteria using the Gene All kit. Subsequently, the plasmid served as a template for VEvhh10 gene amplification using specific primers for the VEvhh10 gene (414bp), Taq DNA polymerase (Figure 2a), and pfu polymerase (Figure 2b). After the amplification process, the PCR product (VEvhh10) was digested with HindIII and XhoI enzymes to create cohesive ends, It has been cleaned (402bp) (Figure 2c). Additionally, the pET-21a vector containing the INP linker was linearized through digestion with HindIII and XhoI restriction enzymes for subsequent pure attachment, the results of this stage were as expected in terms of nucleotide sequence counts.
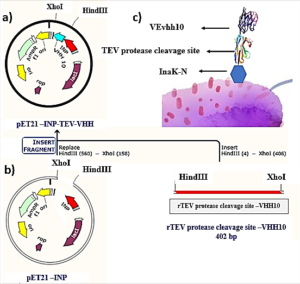

After ligation, the resulting product (pET-21a-Ina -TEV-VEvhh10) was incubated at 4℃ for 14 h and then introduced into bacteria. The transformed product was cultured on ampicillin-containing LB plates. Positive samples were isolated and confirmed for gene insertion using colony PCR. Subsequently, the extracted pET-21a-Ina_K537-TEV-VEvhh10 plasmid was used as a template for amplifying the VEvhh10 gene and the gene structure containing INP. The PCR was performed on the plasmid extraction using Forward and Reverse primers for the VEvhh10 gene 414bp as shown (Figure 3a) and, likewise, Forward and Reverse primers for the T7 promoter and terminator of the plasmid 1099bp as shown (Figure 3b). The plasmid was digested with HindIII and XhoI enzymes, resulting in the visualization of the target gene on the gel (Figure 3c). After examination, the results indicated the success and confirmation of the cloning.
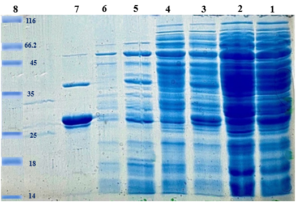
Confirmation of Surface Expression Ina_K537-TEV-VEvhh10
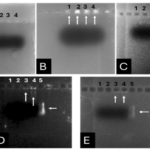
The InaK-N_TEV Protease_Cleavage_Site_VEvhh10 structure consists of three parts: the initial 537 bp of the InaK gene from Pseudomonas syringae encoding a protein with an approximate molecular weight of 19-KD, the VEvhh10 mono body gene with 372 bp and a protein with an approximate molecular weight of 14 KD, and the coding sequence for the TEV protease cleavage site with 21bp. Therefore, the molecular weight of the unmodified InaK-N_TEV Protease_Cleavage_Site_VEvhh10 structure is around 33 KD. To investigate the surface expression of VHH, bacterial pellets were divided into three parts after expression. The first part underwent sonication, the second part was digested with lysozyme, and the third part remained as intact cells (Figure 4). Each of these fractions was also treated with the TEV protease, and the results obtained in SDS-PAGE non-denaturing gel are shown (Figure 4). The appearance of a band in the 33-KD range in SDS-PAGE for the lysed bacterial pellet induced with IPTG, in comparison to the control sample, indicates the expression of this protein. Bands in lanes 1, 3, and 5 represent the expressed surface protein in the presence of TEV protease, indicating a weak TEV protease band and an additional 19-KD band corresponding to INP. In contrast, lanes 2, 4, and 6 show bands corresponding to the expressed surface protein in the absence of TEV protease, and these bands are stronger compared to the previous condition. Additionally, due to the lack of cleavage by TEV protease, the band corresponding to INP did not appear. These results indicate the successful surface expression of VEvhh10 (Figure 5).
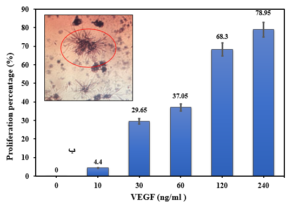
In vitro HUVECs proliferation assay
Before investigating the binding capability of the expressed VHH on the bacterial surface, the activity of the produced non-fused VEGF8-109 was assessed by its effect on the growth and proliferation of human umbilical vein endothelial cells (HUVECs) using the MTT assay. As illustrated in Figure 6, cell proliferation is well-performed with an increase in the concentration of non-fused VEGF8-109. At a concentration of 240 ng/mL, the cell population has reached approximately 80% compared to the sample lacking VEGF8-109. Therefore, the produced non-fused VEGF exhibits biological activity and can be utilized in VHH binding assays.
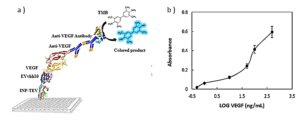
Evaluation of binding of VHHs of VEGF
In order to assess the binding capability of VEvhh10 to VEGF, an ELISA-based immune assay was employed following the mentioned method (Figure 7a). The results obtained from the dose-response curve of VEvhh10 expressed on the bacterial surface using the ELISA method (Figure 7b) demonstrated that an increase in VEGF concentration led to a higher number of anti-VEGF molecules binding to it, resulting in an enhanced optical absorption.
DISCUSSION
According to global statistics, cancer is one of the problems that the world community is facing (34). One promising approach is to inhibit angiogenesis, the formation of new blood vessels, which tumors rely on for growth. Tumor cells can undergo cell death due to oxygen and nutrient deficiency if new vascular systems do not develop. However, when the vascular system extends towards the tumor, it can continue growing. Since angiogenesis is a common requirement across many cancers, targeting it is an effective strategy for tumor elimination. Tumor cells promote vascular growth by secreting VEGF, which affects endothelial cells, triggers signaling cascades, and increases cell proliferation, migration, and survival (35, 36).Vascular endothelial growth factor (VEGF) is the key inducer of angiogenesis, which is crucial for tumor growth and metastasis. Under pathological conditions, VEGF expression significantly increases, with 60% of cancer cells upregulating VEGF to facilitate growth. VEGF promotes endothelial cell proliferation and migration, and increases vascular permeability and protease expression, aiding angiogenesis (10). Due to its pivotal role, VEGF is a primary therapeutic target. Inhibiting VEGF involves strategies like monoclonal antibodies, VEGF mutants, receptor antagonists, soluble receptors, tyrosine kinase inhibitors, anti-sense methods, and aptamers. VEGF functions by binding to its tyrosine kinase receptors (VEGFR-I and VEGFR-II), leading to receptor dimerization and phosphorylation (5). In recent years, targeted therapy using monoclonal antibodies has gained significant attention and emerged as one of the most successful strategies for treating hematologic malignancies and solid tumors. Monoclonal antibodies eliminate tumor cells through various mechanisms, including direct impacts on tumor cells (e.g., blocking receptors), immune system-mediated cell-killing, and specific impacts on tumor angiogenesis (37). While monoclonal antibodies have shown promise in targeted cancer therapy (16, 17), they also come with drawbacks like immunogenicity, high production costs, and limited tumor tissue penetration due to their large size. Effective cancer treatment requires specific properties in monoclonal antibodies, including specificity, solubility, stability, and smaller size. Efforts have focused on producing antibody fragments like scFV and Fab, which offer advantages such as enhanced tumor penetration, reduced immunogenicity, and faster production. However, challenges remain in terms of stability, expression yield, aggregation, and protease resistance, necessitating further improvements (38). In the camelid family, unique immunoglobulins lacking light chains are found in the mammalian immune system. These heavy-chain antibodies feature a VHH domain, which maintains antigen-binding ability akin to full antibodies while overcoming the challenges of conventional antibodies and engineered fragments. Their exceptional characteristics include high stability and solubility, ease of production, small size, heat, detergent, and protease resistance, high homology with human VHH fragments, high antigen affinity, ease of humanization, and engineering into multi-specific forms. Due to their small size, VHHs are expected to penetrate tumors and the retina more effectively than other antibodies. Moreover, VHHs are the only option among antibodies and their derivatives capable of crossing the blood-brain barrier. Therefore, VHHs with VEGF inhibition potential hold promise for angiogenesis inhibition in brain tumors as well (39, 40). Given the extraordinary importance of angiogenesis inhibition in cancer and other diseases, the remarkable success of monoclonal antibodies against VEGF in this field and the superiority of VHHs over other antibodies and antigen-binding fragments have been established. In this study, a surface display system using the INP linker was employed to produce VHH antibodies with high specificity and affinity against VEGF in the bacterial system. The use of a surface display system has various advantages, including low cost, easy expression, and ease of control over expression conditions (41). In this study, we employed a surface display system using the INP linker to produce VHH antibodies targeting VEGF in bacteria, addressing the challenges associated with bacterial cytoplasmic expression (40). The INP-based system offers advantages such as low cost, easy expression, and control over expression conditions. Additionally, the central repetitive region in INP can be removed to produce shorter fusion proteins for cell surface display (42). Despite size limitations in some systems, INP allows expression of proteins up to 60 kD. Various variants of INP exist, with Ina-K being the most commonly used (43), so in this study, the N-terminal region of InaK, which contains the first 537 amino acid pairs of Ina-K, was employed as an anchor motif for displaying VEvhh10 on the surface of bacterial cells. In a 2015 study, Shahangian et al. designed and constructed phages displaying single-domain antibodies targeting the region linked to the VEGF receptor. These phages exhibited binding capability to key functional regions. A phage display library on a nanobody platform was prepared, followed by enrichment and competitive screening of VHHs targeting VEGFR-II using competitive ELISA. Monoclonal VHHs with the highest affinity for the second binding domain of the VEGF receptor were sequenced and named VEvhh1, VEvhh2, and VEvhh3 (VEvhh10) based on their recurrence frequency. The gene sequences encoding these VHHs are deposited in GenBank with accession numbers LC010467, LC010468, and LC010469, respectivel(19).The results of this research indicate that the use of the surface display system with the INP linker for expressing VEvhh10 is a suitable option. This system completely eliminates the need for cell lysis and time-consuming, costly chromatographic methods. With this system, it is possible to express VEvhh10 as shown (Figure 5). Furthermore, the ELISA results and the cell proliferation assay (live-cell assay) confirm that VEvhh10 binds with high affinity to the VEGF receptor binding region, as shown in the (Figure 7.b). Ultimately, a previously unexplored INP, InaA, was successfully used to display VEvhh10 on the cell surface of E. coli BL21 (DE3) (Figure 1.c). The InaK-N_TEV Protease_Cleavage_Site_ VEvhh10 detected on SDS-PAGE was 33 kD, exactly as expected. The expression of VEvhh10 on the bacterial cell surface was verified using the INP Linker. An immunological examination was conducted using an ELISA-based immune assay to measure VEGF concentration in the presence of different VEGF concentrations. As indicated in (Figure 7.b), the results demonstrated that increasing VEGF concentration led to a higher number of anti-VEGF molecules binding (VEvhh10 expressed on the bacterial cell surface), resulting in enhanced optical absorption. This study shows that it is possible to overcome the problems of cytoplasmic expression in bacteria by using a surface expression system. This system transfers VEvhh10 to be expressed on the bacterial cell surface using a linker INP, thus overcoming the problem of incorrect folding inside the bacteria. The information obtained from this study introduces VEvhh10 as a suitable candidate for inhibiting VEGF. It has the capability to block the key functional site of VEGF, inhibiting its binding to its receptor and consequently preventing the cascade of its signaling. However, further studies and investigations are required to validate this proposal.
Acknowledgment
The Researcher expresses sincere gratitude to the Research Deputy of Tarbiat Modares University for providing laboratory facilities and financial support.




![Figure 2: CAPE network architecture [9]](https://journal.hcsr.gov.sy/wp-content/uploads/2024/06/F2-1-300x93.png)
![Figure 3: ClothCap approach [28]](https://journal.hcsr.gov.sy/wp-content/uploads/2024/06/F3-300x192.png)
![Figure 4: Displacement maps with a UV parametrization to represent surface geometry [25]](https://journal.hcsr.gov.sy/wp-content/uploads/2024/06/F4-300x53.png)
![Figure 5: Inria Dataset approach [14]](https://journal.hcsr.gov.sy/wp-content/uploads/2024/06/F5.png)
![Figure 6: Qualitative pose estimation results on BUFF dataset [15] Left to right: scan, Yang et al. [27], BUFF result](https://journal.hcsr.gov.sy/wp-content/uploads/2024/06/F6-214x300.png)
![Figure 7: Annotations of the 3D People Dataset [10]](https://journal.hcsr.gov.sy/wp-content/uploads/2024/06/F7-300x81.png)
![Figure 8: CAPE model for clothed humans [9]](https://journal.hcsr.gov.sy/wp-content/uploads/2024/06/F8-300x100.png)
![Figure 9: Qualitative results on fashion images [9]SMPL [8] results are shown in green, CAPE results are in blue](https://journal.hcsr.gov.sy/wp-content/uploads/2024/06/F9-300x249.png)